
Demis Hassabis
Google DeepMind

John Jumper
Google DeepMind
For the invention of AlphaFold, a revolutionary technology for predicting the three-dimensional structure of proteins
The 2023 Albert Lasker Basic Medical Research Award honors two scientists for the invention of AlphaFold, the artificial intelligence (AI) system that solved the long-standing challenge of predicting the three-dimensional structure of proteins from the one-dimensional sequence of their amino acids. With brilliant ideas, intensive efforts, and supreme engineering, Demis Hassabis and John Jumper (both of Google DeepMind, London) led the AlphaFold team and propelled structure prediction to an unprecedented level of accuracy and speed. This transformational method is rapidly advancing our understanding of fundamental biological processes and facilitating drug design.
A daunting problem
The body’s proteins execute a plethora of vital roles inside cells. Their diverse capabilities are intimately linked to the forms that they take after they fold from linear amino-acid chains into three dimensions. Insights into structure can illuminate function and unlock biological mysteries.
More than 60 years ago, the late Christian Anfinsen (National Institutes of Health) showed that an unfurled protein could regain its shape unaided and concluded that its amino-acid sequence encodes its final organization. As a nascent chain configures itself, it cannot try every possibility: Sampling all arrangements would take longer than the age of the universe, even for a modest-sized protein. Yet inside cells, folding can occur within milliseconds, so nature somehow deciphers the problem. In theory, at least, scientists could discern the guidelines that steer amino-acid chains into correct conformations.
Using multiple approaches, hordes of investigators forged tactics that they hoped would capture this information well enough to mold a protein’s architecture from its sequence. They attempted to express physical interactions in energy equations and looked toward X-ray crystallography and, eventually, other methods to produce templates that could serve as blueprints for related proteins. They also combined knowledge about the chemical proclivities of specific amino acids—whether they carry a charge, for instance—with their location along a chain to gain hints about a protein’s structural features.
In 1994, John Moult (University of Maryland) and colleagues began to track structure-prediction progress by assessing techniques through a community enterprise called Critical Assessment of Structure Prediction (CASP). Every two years, participants receive amino-acid sequences of proteins whose structures have been worked out in the lab, but not yet released. Contestants apply the system that they developed and generate predictions. Those models are then compared with the experimental answer and scored.
Over decades, performance plodded upward in small increments, sometimes stalling or even backtracking. One of the early approaches was developed by David Baker (University of Washington), who used short segments from a shared worldwide database called the Protein Data Bank (PDB) to predict local architecture within proteins. Although helpful in some cases, this fragment-based strategy was time consuming and had limited applicability for the vast majority of proteins.
In the meantime, experimentally solved structures accumulated. In 2014, the number of listings in the PDB surpassed 100,000—but that was still a tiny fraction of the tens of millions of protein sequences available at that time.
Bringing AI into the fold
By 2018 and the 13th CASP competition, scientists had introduced machine learning into their prediction schemes. In contrast to traditional AI approaches that rely on pre-conceived logic, machine-learning systems discover patterns for themselves from data. By making machine learning the central component of their protein-structure prediction network, Hassabis and Jumper’s team won CASP13 with a hefty lead in accuracy over the runner-up and almost a 50% improvement since the best of CASP12. Despite this success, the DeepMind researchers were unsatisfied: They wanted a tool that experimentalists would find useful, with errors of less than one angstrom, the size of an atom.
Hassabis, Jumper, and the AlphaFold team started over and brainstormed intensively. They added geometric and genetic concepts, and they integrated established wisdom about proteins. Atoms have characteristic radii, for instance, and bonds have characteristic angles. The group aimed to include these factors in such a way that they didn’t interfere with the system’s power to learn for itself.
The researchers devised ways to extract maximal information from limited experimental data, and they deployed strategies that force AlphaFold2 to learn efficiently. They allowed the network to adjust calculations anywhere in its process—all the way back to the beginning—as it toils. This innovation avoided a previous pitfall of locking in early errors. Throughout, the system iteratively hones its developing structural model by re-feeding itself tentative solutions.
Hassabis, Jumper, and colleagues also discarded principles that had guided traditional algorithms. For example, they ignored linear proximity in favor of three-dimensional relationships, as amino acids that lie hundreds of subunits away can reside together in a folded protein. Moreover, the team boosted the importance of physical closeness by inventing a mechanism that pays special attention to amino acids that are in contact.
No single element was decisive on its own; rather, many ingenious new ideas combined to achieve a breakthrough performance.
AlphaFold2 takes shape
AlphaFold2 starts with a sequence and crawls databases to find similar ones. It lays out these evolutionary family members as strings of amino acids, one atop the others. It also creates a matrix of information about every possible pair of amino acids within the protein of interest, beginning with their identities, their linear distance, and which direction they lie relative to one another in the chain. These two data sets—the multisequence alignment (MSA) and the pair representations—are processed in parallel during the first stage of AlphaFold2, called the Evoformer. If structures for related proteins have been determined, the system can use those too. Early on, the Evoformer develops a crude structural hypothesis, which it tests and refines. At every step, it tries to fit its model to what it has figured out thus far.
Evoformer measures, for instance, distances between each pair among a set of three amino acids and then assesses whether they form a triangle whose sides meet. If not, this impossibility will eventually need to be resolved, but the discrepancy can be put aside and revisited. In this way, geometry inspires but does not constrain the Evoformer’s activities.
AlphaFold2 incorporates an especially powerful innovation that allows the MSA track, which reflects evolutionary relationships, to communicate with the pairwise representation track, which reflects spatial relationships. As information flows, each path can leverage knowledge that the other one has gained and thus sharpen its own work.
For instance, if the MSA track identifies two amino acids that don’t change over the course of evolution or that co-vary, it can alert the pair-representation track that these amino acids might physically interact. Conversely, if the pair-representation track homes in on possible amino-acid neighbors, it can tell the MSA, which can check whether analogous amino acids in related proteins have coevolved in ways that support that scenario. In this way, crosstalk between the two tracks helps each improve its hypotheses.
After the Evoformer decrypts as many portions of the structure as it can, it passes them to the so-called structural module, which assembles them into a coherent three-dimensional protein. As the structural module jiggles the pieces around, they continue to morph. Initially, it gives every amino acid a position and an orientation that builds a nonsensical conglomeration: They all lie in the same place.
Step by step, it rotates and moves the amino acids, still ignoring which ones are linearly adjacent. Eventually, the protein’s backbone emerges and the system places the chemical side chains that characterize each amino acid. AlphaFold2 predicts not only the entire 3D structure, but also reliability scores for each part.
Rigorous training
To train the system, Hassabis and Jumper’s team used the PDB’s experimentally established structures. AlphaFold2 repeatedly compared its proposals to the real answer and gradually nudged its solutions closer to reality. By repeating this process on every member of the training set, the algorithm absorbed principles of protein structure.
The researchers exploited tricks that pushed the network to learn better. For instance, they hid amino acids in the MSA and asked it to fill in the gaps. In this way, they demanded that the system master rules of evolutionary relationships. They also recursively supplied outputs from any given step, which provided many opportunities for AlphaFold2 to reconsider and refine.
AlphaFold2 also computed how much to trust its predictions, and these confidence ratings allowed the researchers to squeeze more information from the available data and thus, to enhance its performance. After they fed it the roughly 140,000 PDB sequences, they ran another set whose structures had not been solved. From the predictions, they plucked the most reliable 350,000 sequence/structure pairs and trained the system on those data as if they had been experimentally verified.
Retooling protein science
In 2020, AlphaFold2 soared past the competition in CASP14. Its predictions were accurate to atomic precision and it generated excellent results in minutes even for proteins that lacked a template. This was the first approach that could construct high-resolution predictions in cases where no similar structure is known.
In July 2021, Hassabis and Jumper published their method as well as structure predictions of almost every human protein. In only two years, their manuscript’s impact has vaulted over almost all of the 100,000 research articles that have been published in Nature since 1900. It ranks 50th, having been cited in more than 7000 papers from top journals.
In collaboration with the European Molecular Biology Laboratory’s European Bioinformatics Institute, Hassabis and Jumper have shared the program and the database with the scientific community, and more than a million investigators have used these resources. The DeepMind team has since expanded its catalog to almost every known protein in organisms whose genomes have been sequenced. Listings include proteomes of, for instance, viruses that pose epidemic threats and the World Health Organization’s high-priority pathogens.
The technology has already made a dramatic impact in myriad biomedical spheres and beyond. It helped researchers fill holes in their visualization of the nuclear pore complex, an enormous and complicated molecular machine that controls transport into and out of the nucleus. Scientists used the tool to analyze a bacterial syringe that shoots molecules into insect cells. By applying understandings that AlphaFold2 revealed, the investigators reengineered the protein to target human cells, opening a new avenue toward medication delivery and gene therapy. Academic laboratories and companies are harnessing AlphaFold2 to develop vaccines, design drugs, craft enzymes that chew up pollutants, and much more. The prospects are endless.
By letting their imaginations and talents fly, Hassabis, Jumper, and their team completed a quest that had flummoxed scientists for half a century. This triumph has launched a new era in studying and manipulating proteins. It has already catalyzed substantial advances, and its impact and reach promise to explode as workers in a vast range of fields dream up new ways to mine its potential.
by Evelyn Strauss
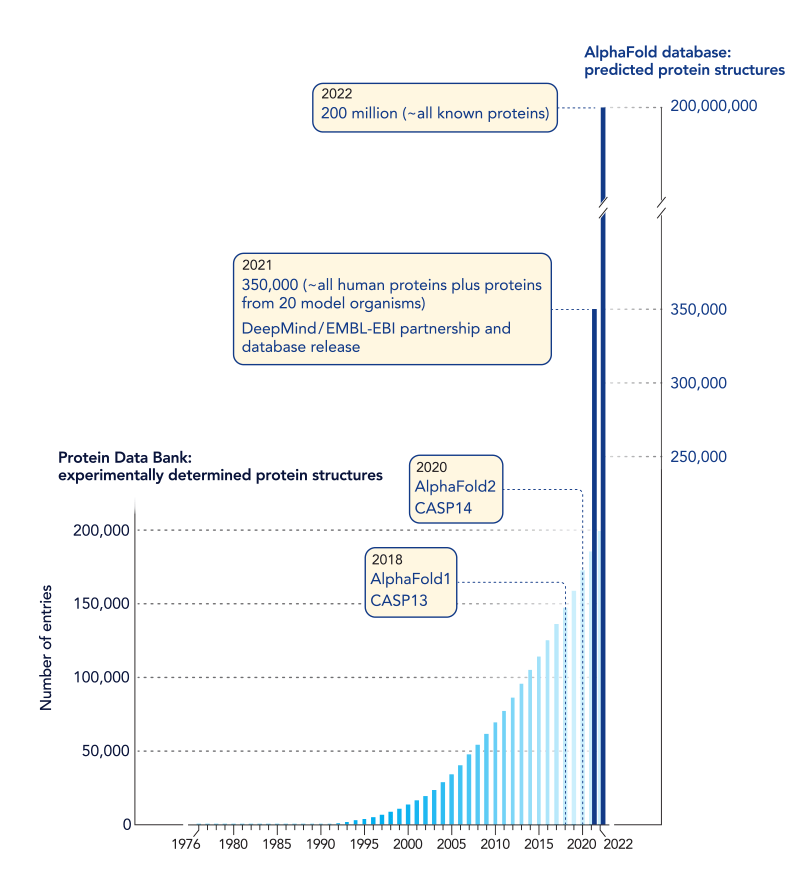
Predicting success
Beginning in 1957, when John Kendrew first determined the three-dimensional shape of a protein, scientists have made slow and steady progress on solving protein structures by experimental means. In the last several years, machine learning has catapulted the protein-structure field into a new realm. By 2022, a year after the DeepMind team introduced AlphaFold2, the group had generated predicted structures for almost all known proteins—approximately 200 million of them—an increase of three orders of magnitude over the total number of experimentally solved structures.
EMBL-EBI = European Molecular Biology Laboratory-European Bioinformatics Institute
CASP = Critical Assessment of Structure Prediction
Selected Publications – Demis Hassabis and John Jumper
Senior, A.W., Evans, R., Jumper, J …..…. Kavukcuoglu, K. and Hassabis, D. (2019). Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins: Structure Function and Bioinformatics. 87, 1141-1148.
Senior, A.W., Evans, R., Jumper, J …..…. Kavukcuoglu, K. and Hassabis, D. (2020). Improved protein structure prediction using potentials from deep learning. Nature. 577, 706-710.
Tunyasuvunakool, K., Adler, J …..…. Jumper, J. and Hassabis, D. (2021) Highly accurate protein structure prediction for the human proteome. Nature. 596, 590-596.
Jumper, J., Evans, R …..…. Kohli, P. and Hassabis, D. (2021). Applying and improving AlphaFold at CASP14. Proteins: Structure Function and Bioinformatics. 89, 1711-1721.
Jumper, J., Evans, R., Pritzel, A …..…. Kohli, P. and Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature. 596, 583-589.
Jumper, J. and Hassabis, D. (2022). Protein structure predictions to atomic accuracy with AlphaFold. Nature Meth. 19, 11-26.

The Secret to a Successful Career in Science—According to Magritte
There is no shortage of words of advice on how to become a successful scientist.
Award Presentation: Erin O’Shea
Cells in our body rely on proteins to perform many different tasks, including digestion of our food, contraction of our muscles, and transmission of signals between neurons inside our brain. Proteins are made up of building blocks called amino acids that are joined to each other in a particular order like different colored beads on a string. The order of amino acids is unique to each protein and is specified by our DNA. For proteins to carry out their functions, they must fold into a three-dimensional shape in which some amino acids that are far from one another in linear sequence come close to each other when the protein adopts its final shape. The process by which a protein goes from beads on a linear string to a three-dimensional structure is called protein folding.
For many years scientists have worked to understand how proteins fold into their structures. What tells a protein how to fold properly? Are there machines inside cells that help proteins fold? Or is the information that specifies the folded structure of a given protein contained within the sequence of amino acids itself? Part of the answer to these questions came from a beautiful experiment that Christian Anfinsen performed in 1962 (ref. 1). He put a protein into a test tube and used chemicals to unfold it – to destroy its folded structure but to leave the sequence of amino acids intact. He then asked what would happen if he removed the chemicals that caused the protein to unfold – would the protein fold back into its characteristic structure or not? Anfinsen got a clear and surprising result from this experiment – when he removed the unfolding chemical, the protein returned to its characteristic shape. This told Anfinsen that, at least for the protein he was studying, the information that specifies what structure the protein will adopt is contained in the sequence of amino acids. For this work Anfinsen was awarded the Nobel Prize in 1972.
After Anfinsen’s experiment, the race was on to learn how to predict the folded structure of a protein from its sequence of amino acids. If this could be done, we might better understand the jobs that proteins perform inside of cells, learn how mutations cause disease, and more easily identify drugs that are able to interact with proteins and thereby treat diseases. Over the years scientists have labored to determine protein sequences and protein structures. This has been a time-consuming, costly, and labor-intensive process—one which has been able to reveal the structure of only a mere fraction of known proteins. From this work, scientists have learned that proteins with similar sequences of amino acids adopt similar folded structures, reinforcing the notion that there is a code relating protein sequence to structure.
In 1994 a community-based competition called CASP (which stands for Critical Assessment of Structure Prediction) was launched to identify the algorithms that could most accurately predict protein structure from protein sequence. In each annual competition, scientists are given sequences of test proteins whose structures are not known and are challenged to use computer-based methods to predict their structures. While the competition is happening, other scientists use experimental approaches to determine the structures of these test proteins. The winner is the scientist or group that can most accurately predict protein structures from protein sequences. For decades, scientists have made steady progress. But in the 2020 CASP competition, that all changed (ref. 2). The group at Google DeepMind led by John Jumper and Demis Hassabis generated structure predictions that were several-fold better than the other groups in the competition. In fact, many of the DeepMind predictions had accuracy comparable to those determined by experimental methods. After 30 years of competitions, the judges who assessed the model predictions declared the 50-year-old Anfinsen problem solved – most protein structures can be predicted from their sequences.
The team led by Jumper and Hassabis used their model to predict structures for more than 200 million protein sequences, which is already changing the practice of biology in labs worldwide.
So how did DeepMind do this? The short answer is that they built a computational model that relies on artificial intelligence or AI (ref. 3). But this is an oversimplification, as DeepMind was not the first to use AI approaches to address the protein structure prediction problem. So what made DeepMind’s approach so successful (ref. 4)? First, AI approaches make use of large sets of data to “train” the computational model, and in recent years we have seen an explosion in the amount of protein sequence and protein structure data. Second, DeepMind’s success relied on a combination of creative ideas, brilliant engineering, and powerful computers. The DeepMind algorithm, termed AlphaFold2, consists of several modules – interlinked computational building blocks that each perform a task. The algorithm starts with a module that searches databases with the protein sequence of interest, generating an alignment that consists of similar but not identical sequences from different organisms. Using these alignments, amino acids that change throughout evolution can be identified, and then correlations can be found between changing amino acids, which others have shown are typically in contact with one another in the folded structure. Another module searches the database of protein structures for those that are similar to the sequence of interest and generates a representation of which amino acids might be in contact with one another, called a contact map. What happens next is part of the secret sauce of DeepMind’s approach. The outputs from the previous two modules are fed into another part of the algorithm that determines which pieces of information are the most valuable, seeking to improve both the sequence alignment and the contact map. After several cycles of iteration, the refined sequence alignment and contact map are fed into a final module that constructs a three-dimensional representation of the protein. A critical feature of the DeepMind approach is that it is iterative – the final structure is fed back into the previous modules to refine the sequence alignment and contact map, which are used to produce a refined structure.
So why is the ability to predict protein structure so important? First, knowledge of the shape of a protein – its structure – many times reveals things about its function inside of cells. Second, modern drug discovery relies heavily on knowledge of protein structure, and being able to predict structure accurately from sequence promises to speed up this process. We can see how drugs fit into binding pockets and design new drugs once we understand what parts of the protein are interacting. Third, being able to predict protein structure helps us understand how disease-associated mutations might affect protein structure and function, paving the way to better understand disease and how to treat it. And finally, there is a lot of interest in designing proteins that have new functions – for example, proteins that can bind to the receptor that the SARS-CoV2 virus uses to get into cells, thereby preventing the virus from gaining access. The ability to predict structure from sequence will likely speed up the process of protein design.
For this landmark discovery, John Jumper and Demis Hassabis are being awarded the 2023 Albert Lasker Basic Medical Research Award.
References:
(1) Haber, E. and Anfinsen, C. B., J. Biol. Chem. 237, 1839 (1962).
(2) Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Proteins 89, 1607-1617 (2021).
(3) Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S. A. A.; Ballard, A. J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A. W.; Kavukcuoglu, K.;Kohli, P.; Hassabis, D. Nature 596, 583−589 (2021).
(4) AlphaFold 2 is here: what’s behind the structure prediction miracle<
Acceptance remarks
Acceptance Remarks: Demis Hassabis
I am deeply honoured to receive the 2023 Lasker Basic Medical Research Award together with John Jumper. In common with almost any research breakthrough in modern science, the work is the result of a large multidisciplinary team effort and we would like to acknowledge the immense contributions of our wonderful colleagues on the AlphaFold team and also more widely across the whole of DeepMind and Google.
The journey we’ve been on has been amazing. I remember first coming across the protein folding problem 30-years ago as an undergraduate, and becoming fascinated by it, thinking how it might be the perfect type of problem to tackle with AI one day.
When we founded DeepMind in 2010, our goal was to use artificial intelligence to advance knowledge and accelerate scientific discovery. The watershed moment came in 2016 when our prior system, AlphaGo, became the first AI program to beat a world champion at the complex and ancient game of Go. I knew then we had the general AI knowhow and ideas to tackle as formidable a challenge as protein folding and the AlphaFold project was born.
It took many further years of development and innovation specific to the problem of course. AlphaFold proved to be the most difficult and complex AI system we had ever built, but when we received the results for the CASP13 competition in 2020 we knew we had achieved atomic accuracy on the target protein structures, an amazing moment that none of us on the team will ever forget.
Since AlphaFold’s release, we have been thrilled by what the scientific community has done with it. To date it has been used by over a million researchers to advance a huge and diverse range of work, everything from enzyme design to disease understanding to drug discovery. The speed with which AlphaFold has been adopted by the biological community as a standard research tool has been very gratifying to see – it is everything we hoped for and more, and hopefully just the beginning of the impact it will make.
AlphaFold is one of the first examples of what I like to call ‘science at digital speed’, both in terms of the speed of the solution (a few seconds for an average protein) and the speed of the dissemination of that solution (as fast as a keyword search on a database). We very much hope that when we look back on AlphaFold in a decade’s time it won’t just be an isolated instance of AI being productively applied to modelling complex biological phenomena but in fact the heralding of a new golden era of digital biology.
