
Stephen J. Elledge
Brigham and Women’s Hospital

Evelyn M. Witkin
Rutgers University
The 2015 Albert Lasker Basic Medical Research Award honors two scientists for their discoveries concerning the DNA-damage response, a mechanism that protects the genomes of all living organisms. Evelyn M. Witkin (Rutgers University) established its existence and basic features in bacteria, and Stephen J. Elledge (Brigham and Women’s Hospital) uncovered its molecular pathway in more complex organisms. The details of the two systems differ dramatically, yet they share an overarching principle. Both coordinate the activity of a large number of genes whose products shield creatures from potentially lethal harm.
Throughout their lives, cells withstand an onslaught of insults to their DNA. External agents such as chemicals and radiation bash it, and it also sustains abuse from within when normal physiological processes blunder. In humans, such events deliver tens of thousands of genetic wounds every day. The DNA-damage response detects not only DNA anomalies, but also other dangers, such as interruptions in the DNA-copying process. Living creatures then implement a multi-pronged strategy to ensure survival.
Like bacteria, mammalian cells construct DNA-repair equipment and arrest division when they detect genetic peril. In addition, when the extent of injury overwhelms DNA-restorative capacities, the DNA-damage response sparks cell suicide. The organism thus maintains quality control and defends itself against cancer, an illness that is characterized by rampant genetic misconduct, including unbridled duplication of cells that carry marred DNA. Flaws in the human version of the DNA-damage response system cause diverse illnesses, including cancer, neurodegenerative disorders, and immune deficiencies.
Radiating insights
When Evelyn Witkin began studying the basis of radiation resistance at Cold Spring Harbor Laboratory in 1944, researchers had barely established that DNA was the genetic material. Radiation in the form of X-rays and ultraviolet (UV) light was known to cause inherited genetic changes, but the molecular nature of such mutations remained obscure, as did the method by which cells guard against them.
In her first experiment with bacteria, Witkin inadvertently uncovered naturally occurring Escherichia coli variants that resist radiation’s ill effects. She intended to induce mutations, but she had no experience with the techniques and used a UV dose that was so high, it killed almost all the microbes. Of the 50,000 cells that she put on her petri dishes, four grew. They had somehow overcome the parent bacterium’s unusual radiation sensitivity.
To divide successfully, a bacterium must create a partition that separates the two daughter cells, and UV exposure temporarily impedes this process. The parental strain that Witkin used in her studies exaggerates the usually brief delay that is typical of most bacteria. In the parent strain, she found, tiny amounts of UV light demolished the microbe’s ability to create the partition, and the creature never recovered. Consequently, long spaghetti-like filaments developed and the cells eventually died. The UV-resistant strain, in contrast, resumed division after a short lag.
Alert to SOS
In the 1960s, Witkin began uniting these observations with others. First, she noticed parallels between two previously unrelated behaviors. In addition to evoking filamentous growth of bacteria, UV awakens bacterial viruses called phages, whose DNA has settled silently into the bacterial genome. Other investigators had just shown that UV irradiation initiates phage activation by destroying a protein that normally restrains its genes.
By that point, Witkin had discerned that UV treatment of her parent E. coli strain stimulates production of a substance that hinders cell separation. Perhaps, she speculated, manufacture of this substance normally is limited by an inhibitor (commonly called a repressor) that resembles the one that hampers phage gene activation. Maybe the same UV-induced molecular apparatus incapacitates both of the inhibitors, thus prompting filamentous growth and phage activation. This idea gained support from another group’s observation that cells with a single genetic defect spur both processes. A common pathway seemed to link the two phenomena.
In the meantime, Witkin had generated key insights into the mechanism by which UV light causes mutations. Radiation by itself does not generate inherited changes; subsequent cellular functions are required, she found. UV light triggers chemical reactions within DNA molecules that disrupt its structure and render the genetic code uninterpretable at those spots. Unless the original DNA letter is restored, she proposed, a promiscuous DNA-replication machine — an enzyme that is sloppier than the only one known at the time, which stops cold when it encounters DNA damage — inserts a random, and often incorrect, DNA building block. Witkin thus predicted accurately that DNA damage stirs production of an error-prone copying enzyme that fosters mutagenesis long before there was direct evidence for it.
Furthermore, UV mutagenesis in E. coli requires a protein called LexA. Perhaps, she suggested in 1967, LexA normally limits production of the error-prone enzyme. Two years later, she implicated a second protein, RecA, in generation of UV-induced mutations.
In the early 1970s, Miroslav Radman and colleagues (Free University of Brussels) found that another UV-induced process—Weigle mutagenesis—also requires LexA and RecA, and they pointed out that phage awakening shares these features. Radman subsequently suggested that these phenomena and possibly UV-induced bacterial mutagenesis depend on error-prone DNA replication, which he called SOS replication in reference to the universal distress signal. Witkin soon established experimentally that UV-induced bacterial mutagenesis belongs to this cluster of SOS activities.
Invigorated by this growing constellation of commonly controlled behaviors, she and Radman scoured the scientific literature for more examples of UV-inducible functions whose activities depend on RecA and LexA. We now know that the SOS response (see Figure) switches on dozens of genes whose products contribute to a broad array of activities, including DNA repair and mutagenesis, that promote survival under stressful circumstances.
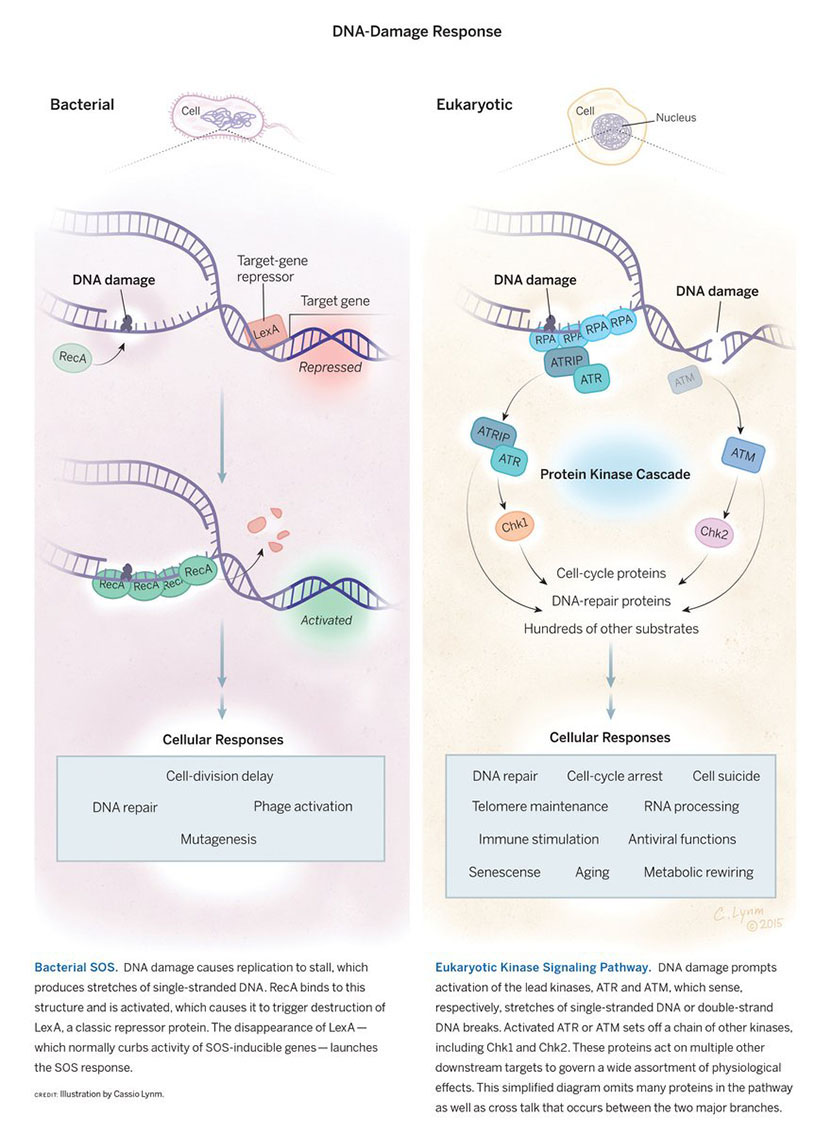
From yeast to humans
By the late 1980s, scientists knew that eukaryotes, whose cells (unlike bacteria) contain nuclei, also respond to DNA damage. This system’s mechanism, however, was opaque. In 1987, Stephen Elledge and his postdoctoral advisor, Ronald Davis (Stanford University School of Medicine), accidentally discovered that quantities of the messenger RNA for a yeast subunit of an enzyme called ribonucleotide reductase soar when DNA is damaged and replication is blocked. That behavior made sense, as the enzyme helps construct DNA building blocks, raw materials that are needed for DNA repair and synthesis. This observation and others inspired Elledge to suggest that a signaling system detects deviant DNA structures and, in turn, adjusts the activity of multiple genes whose products contribute to DNA synthesis and repair. Conventional wisdom held that triggers for signaling pathways had to come from outside rather than inside cells, and his bold idea was not initially accepted.
With his own students and postdoctoral fellows (initially at the Baylor College of Medicine and subsequently at Harvard), Elledge devised numerous genetic tricks to investigate his idea. By hooking up the genetic control region for a ribonucleotide reductase subunit to a reporter that indicates when it is active, Elledge identified yeast strains that carry altered versions of machinery in the hypothetical pathway. Some provoke DNA-damage-inducible genes in the absence of DNA damage, whereas others fail to rouse these genes in its presence.
The first class of yeast strains included one with a defective version of an enzyme that makes DNA. Because faltering DNA replication in this strain mimics the effects of DNA damage, the observation established that stalls in DNA synthesis can prod damage-stimulated genes.
The second, “DNA damage uninducible,” class of genes, included one (DUN1) that encodes a classic signaling enzyme, a kinase. Kinases activate or inactivate proteins by adding chemical adornments called phosphates to them, and they often fire sequentially, turning one another on and off. Dun1 itself gains phosphates in response to DNA damage, Elledge showed. The protein thus emerged as a strong candidate for participation in the signaling pathway that he had predicted, and thus generated support for its existence. Intact Dun1 is required to incite DNA-damage-inducible genes, has the enzymatic potential to transmit messages to downstream members of the pathway, and is activated in response to DNA damage.
Additional genetic strategies allowed Elledge to place more participants into the yeast pathway. He showed that DNA damage and replication stalls converge on a single protein, another kinase called Sad1/Rad53, that governs Dun1 activity; he also identified two kinases, Mec1 and Tel1, that act upstream of Sad1/Rad53. These proteins provided a potential link to mammals, as they belong to a molecular family that includes a human protein, ATM. Defects in ATM underlie a fatal disease (ataxia telangiectasia) whose symptoms include a high incidence of cancer, and cells from individuals with the illness fail to arrest division appropriately in response to DNA damage.
Elledge and numerous other investigators, including Michael B. Kastan (then at St. Jude, Memphis), Antony M. Carr (University of Sussex), and William G. Dunphy (California Institute of Technology), subsequently worked out key features of the mammalian DNA-damage response pathway and showed that it closely resembles that of yeast. These organisms utilize a series of related kinases to drive activities that protect themselves from threats to genomic integrity (see Figure).
Simple signal, complex effects
Elledge’s illumination of the human pathway revealed in 2001 that ATR, a mammalian ATM-related kinase known to be essential for the DNA-damage response, requires another protein to do its job. The second protein, ATRIP, led him to the mechanism by which the pair senses DNA aberrations.
A molecule called replication protein A (RPA) plays a crucial role in this step, Elledge discovered in 2003. It sticks to single-stranded DNA, a structure that is generated by multiple types of DNA lesions. ATRIP grabs RPA-coated single-stranded DNA, bringing along the ATR to which it is bound. This event kicks off the ATR arm of the DNA-response pathway.
Although numerous participants in this system had emerged, the total collection had never been defined. To probe this issue, Elledge exploited the fact that ATM and ATR add phosphates at known amino acid pairs within their target proteins. In 2007, he identified more than 700 proteins that receive phosphates from ATM or ATR in response to DNA damage. These proteins perform a tremendous range of activities (see Figure) that are crucial for health, and especially for preventing malignancy.
Creatures as diverse as bacteria and humans react to DNA damage by instigating a multifaceted physiological response that enhances their ability to endure the assault and thrive in its aftermath. Witkin and Elledge laid the conceptual and experimental foundation for our understanding of these intricately organized systems, which ensure genetic fidelity and safeguard organismal vitality.
by Evelyn Strauss
Key publications of Stephen Elledge
Elledge, S.J. and Davis, R.W. (1989). Identification of the damage responsive element ofRNR2 and evidence that four distinct cellular factors bind it. Mol. Cell. Biol. 9, 5373-5386.
Zhou, Z. and Elledge, S.J. (1993). DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell. 75, 1119-1127.
Sanchez, Y., Desany, B.A., Jones, W.J., Liu, Q., Wang, B., and Elledge, S.J. (1996). Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 271, 357-360.
Sanchez, Y., Wong, C., Thoma, R.S., Richman, R., Wu, Z., Piwnica-Worms, H., and Elledge, S.J. (1997). Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation via Cdc25. Science. 277, 1497-1501.
Matsuoka, S., Huang, M., and Elledge, S.J. (1998). Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 282, 1893-1897.
Zou, L. and Elledge, S.J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 300, 1542-1548.
Harper, J.W. and Elledge, S.J. (2007). The DNA damage response: ten years after. Mol. Cell. 28, 739-745.
Key publications of Evelyn Witkin
Witkin, E.M. (1947). Genetics of resistance to radiation in Escherichia coli. Genetics. 32, 221-248.
Witkin, E.M. (1967). The radiation sensitivity of Escherichia coli B: a hypothesis relating filament formation and prophage induction. Proc. Natl. Acad. Sci. USA. 57, 1275-1279.
Witkin, E.M. (1969). The mutability toward ultraviolet light of recombination-deficient strains of Escherichia coli. Mutat. Res. 8, 9-14.
Witkin, E.M. (1974). Thermal enhancement of ultraviolet mutability in a tif-1 uvrA derivative of Escherichia coli B/r: evidence that ultraviolet mutagenesis depends upon an inducible function. Proc. Natl. Acad. Sci. USA. 71, 1930-1934.
Witkin, E.M. (1976). Ultraviolet mutagenesis and inducible DNA repair in Escherichia coli. Bacteriol. Rev. 40, 869-907.
Witkin, E.M., (1991). RecA protein in the SOS response: milestones and mysteries. Biochimie. 73, 133-141.
Witkin, E.M. (2002). Chances and choices: Cold Spring Harbor 1944-1955. Annu. Rev. Microbiol. 56, 1-15.
Award presentation by Titia de Lange
The oldest written texts are 5000 years old and are only known today because their Sumerian authors used clay tablets and kiln-baked their texts into permanence. The Etruscans hammered their best ideas onto gold plates, which also survived thousands of years. The progression to papyrus and paper often made written words short-lived and now the transition to electronic text has reduced that fleeting time to a New York minute.
What we write on our iPads is doomed to disappear fast, and baking our tablets in a kiln will not bring permanence. Of course the same changes that shortened the half-life of our texts came with enormous improvements in multiplication and distribution. But we may want to consider some mechanism to enhance the durability of what we write, even if most of it does not seem worth preserving.
Acceptance remarks
Acceptance remarks, 2014 Lasker Awards Ceremony
I got hooked on science in part because of the space race in the 1960s. There was a push to more broadly educate kids in science, and consequently I was exposed to the SRA reading laboratory in school. The advanced material was on science — fossils, planets, atoms and molecules. I loved atoms. The idea that matter was composed of smaller and smaller units with definable properties was astonishing. The fact that elements fell into the periodic table in a way that could predict their chemical properties is an amazingly elegant fact of nature that drew me in and continues to astound me even today. Initially, though, I avoided biology. But my senior year in college I learned about recombinant DNA — biology had gone molecular and that intersected with my interests in chemistry. The more I learned about DNA, the more I became seduced. In my mind, there is no more beautiful piece of art than the double helical structure of DNA. It is beauty and elegance personified. I wanted to be part of that revolution.
I was fortunate to be accepted to MIT without a biology background. I can remember when I first began doing research at MIT. I could not wait to get into the lab look at the results of my genetic experiments — did it work? I was a kid in a candy store and I just wanted to spend all of my time testing new ideas in the lab. It is an indescribable feeling of enlightenment when the right idea clicks into place to explain a phenomenon, an incredible rush. I am especially thrilled to share this award with Evelyn Witkin, whose pioneering work I learned of at MIT. While our careers barely overlapped, our interests were congruent. Can DNA sense its own integrity? What does it do with that information? We were both incredibly fortunate to work on such a rich and rewarding project.
There is nothing I would rather do than to be a scientist. It is a privilege to be able to pluck one of Mother Nature’s problems out of its firmament and examine how it works before putting it back, hopefully as a now shinier object. And to have something good come out of this work, to impact health and society, is more than I could have ever imagined. I am so grateful to have been able to play a role in science.
For me, the Lasker Award is a profound personal honor. I know that it recognizes not just the work of many creative individuals in my lab, but it is also a celebration of science itself. It is important for society to promote the culture of science and celebrate its values, the values of reasoning, openness, tolerance, and respect for evidence. That is why I so value the Lasker and its heritage. Mary Lasker saw far into the future and convinced this country of the importance of science and basic research, making a tremendous impact on society at large. By continuing to celebrate stellar science year after year, the Lasker Foundation is promoting the values of science.
I would like to end with a quote from Albert Einstein:
“One thing I have learned in a long life: That all our science, measured against reality, is primitive and childlike — and yet it is the most precious thing we have.”
Acceptance remarks, 2014 Lasker Awards Ceremony
I am deeply grateful to the Lasker Foundation and the Lasker Jury for the huge honor of this Award, which I am proud to share with Steven Elledge. I admit, though, that it feels slightly wicked to be recognized for work that was a labor of love and has always been its own reward.
John Tyndall, the 19th-century Irish physicist, in describing the mind of the scientist, wrote: “He lives a life of the senses, using his hands, eyes and ears in his experiments, but is constantly being carried beyond the margin of the senses. His mind must realize the subsensible world, and possess a pictorial power; if the picture so formed be correct, the phenomenon he is investigating is accounted for. Imagination with him does not sever itself from the world of fact; this is the storehouse from which all its pictures are drawn…”
I found that John Tyndall’s “pictorial power” always played a part in my scientific style. By 1961, after ten years of investigating the mechanism of ultraviolet light- (UV) induced mutagenesis in Escherichia coli, I had reached a conclusion. My experiments had told me that a UV-induced mutation occurs when an unrepaired UV photoproduct, most likely a pyrimidine dimer, prompts a replication error during the first DNA replication following UV exposure. I published those experiments and that conclusion, but not the vivid images that had formed in my mind, of just how that might happen.
In the 1960s, only one of E. coli’s five DNA copying enzymes, the DNA polymerases, was known, the one we now call Pol I. We also knew that this DNA polymerase was stopped cold by an unrepaired pyrimidine dimer in the DNA. That’s where imagination took over. I could almost ‘see’ another DNA polymerase, less fastidious than Pol I, knocking PoI I off the DNA and taking over as the copying enzyme, inserting any base at random, with a high probability of error, opposite the noncoding UV photoproduct.
In 1967, when I found that a mutation in the lexA gene eliminated UV mutability, I proposed that this gene encodes or controls just such a new DNA polymerase, responsible for UV mutagenesis by performing what we now call error-prone translesion synthesis.
In the early 1970s, Miroslav Radman and I pooled our separate observations and insights and recognized that UV damage induces a cluster of coordinately regulated, DNA damage-inducible activities, which came to be known as the SOS response. In time, dozens of E. coli genes were identified as SOS-inducible, including the umuC and umuD genes, essential for UV mutagenesis. It began to be possible to picture particular proteins carrying out translesion synthesis.
Harrison (Hatch) Echols, before his untimely death in 1983, was engaged in reconstructing SOS mutagenesis in vitro, with purified proteins. Hatch’s project has been carried on by Myron Goodman, Roger Woodgate, and coworkers at UCLA. They have identified the SOS-inducible error-prone DNA polymerase as Pol V, and have shown that it is composed of a particular arrangement of the UmuD and UmuC proteins. Six years ago, they showed that the “mutasome,” the active complex that performs SOS-induced translesion synthesis, is made up of Pol V+ RecA protein +ATP.
By this time, I was enjoying not just a visual image of SOS mutagenesis. It was more like watching an animated video with multiple components shown in such beautiful molecular detail that it was, for me, like seeing a long-imagined dream come true. I’m very glad that I lived to see it, and so sorry that Hatch didn’t.
Interview with Evelyn Witkin and Stephen Elledge
Video Credit: Flora Lichtman
