
Mario Capecchi
University of Utah, Howard Hughes Medical Institute

Martin Evans
Cardiff University

Oliver Smithies
University of North Carolina at Chapel Hill
For the development of a powerful technology for manipulating the mouse genome with exquisite precision, which allows the creation of animal models of human disease.
This year’s Lasker Basic Medical Research Award honors three scientists who developed a powerful technology that allows scientists to create animal models of human disease. With this technology, researchers have engineered mice with conditions such as atherosclerosis, cancer, high blood pressure, and cystic fibrosis, allowing the study of many debilitating disorders. And the same technology is uncovering the secrets of normal biological processes as well, revealing, for example, how the nervous system develops or how immune cells collaborate to quash microbial invaders.
This progress depends on the ability of researchers to manipulate the genetic material of mice with exquisite precision. Scientists can disable — or knock out — a gene, and they can also change its properties in more subtle ways. These procedures generate animals that carry specific genetic alterations, and recreate the underlying cause of a human disorder or uncover the role of any gene of interest.
Inactivating a mouse gene takes two steps. In the first, scientists snip out its middle in a test tube using conventional tools of molecular biology. The challenge then is to replace the intact, functional gene in the mouse chromosome with the modified version created in the test tube. This genetic swap requires the introduced DNA to find the corresponding DNA in the chromosome. Less than two decades ago, conventional wisdom held that this task was impossible in mammalian cells, and that DNA could insert only at random sites.
At the time, knocking out a particular gene was about as successful as tossing a dart into a heap of spaghetti and hoping it hits a particular strand in a certain spot. Scientists could add DNA to mouse cells, but it landed in random places. They couldn’t yet target DNA to a particular site in the chromosome — in other words, to a specific gene. Today, hitting a bull’s-eye is a standard maneuver.
By building on several decades of fundamental studies in mouse embryology and molecular genetics, Mario Capecchi, Martin Evans, and Oliver Smithies brought this work to fruition and delivered the technology of gene targeting in living mice to labs around the world. Scientists can now create and breed mice with particular pathologies, study them systematically, evaluate the functions of genes in an intact mammal, and dissect even the most complex processes.
How cancer helped
The seeds of this technology grew out of a bizarre form of cancer of the gonads, called a teratocarcinoma. Most tumors possess characteristics of their specific cell of origin, but teratocarcinomas display features of many different tissues. Skin, muscle, hair, and even teeth compose these masses. Researchers showed in the 1960s and 1970s that this chaotic mixture of tissue types is all derived from an immature type of cell with many potential fates, called an embryonic carcinoma (EC) cell.
In this respect, EC cells from teratocarcinomas resemble embryonic stem (ES) cells from normal animals, which scientists knew existed, but could not isolate. ES cells develop soon after fertilization, and they, too, have not yet committed to any particular tissue type. A number of labs showed that EC and ES cells are close cousins in other ways as well. Early embryos turn into teratocarcinomas instead of mice if they are implanted into sites in an adult mouse other than the uterus. And most importantly for the enterprise of gene targeting, either type of cell injected into very young embryos grows into normal parts of the fully developed mouse. The resulting ‘chimeric’ animals carry some cells from their ‘normal’ mother and father, and some cells from the mouse that donated the EC or ES cells.
In principle, if scientists could alter the DNA of a cell that would contribute to a live mouse, they could create an animal with any desired genetic change. The process would involve altering the cell’s DNA in a culture dish and generating a chimeric mouse that incorporates that cell’s new genetic information into its sperm and eggs. But EC cells didn’t lend themselves to this task. They contributed to most parts of the mouse, but it proved difficult to produce animals with the EC DNA in egg or sperm, an accomplishment that was essential to breed animals with the genetic change. ES cells seemed more promising, but they presented a different problem. Manipulating genes and then sorting through many cells to find the desired alterations wasn’t possible with these cells because scientists couldn’t grow them in the culture dishes where such maneuvers would take place.
Martin Evans, then at the University of Cambridge and now at Cardiff University in the UK, considered several explanations for the inability to culture ES cells from the early embryo. Such cells could be rare, he reasoned, and they might not survive well outside of the mother. Furthermore, they might exist for only a very short time at an exact phase of embryonic development.
Evans zeroed in on the time period when he thought ES cells existed in the embryo. To increase the number of potential cells, he and his colleague Matt Kaufman delayed implantation of the early embryo into the mother’s uterus. This procedure allowed the embryos to accumulate cells at the right stage.
In the early 1980s, Evans and Kaufman harvested the early embryos, grew them in culture dishes, and picked out cells that closely resembled EC cells. They had hit gold. These cells formed teratocarcinomas when injected into mice. Furthermore they could be maintained for long periods of time in culture and specialized magnificently well in the Petri dish. They possessed all the features that identified them as the sought-after embryonic stem cells.
Evans had isolated the much-pursued multi-purpose cells. In principle, their DNA could be manipulated to create any mutation of interest. But how were scientists going to accomplish this task with the exquisite precision they desired? The only way to manipulate an organism’s DNA at the time was to integrate genes randomly. This ability to produce so-called transgenic mice has proved extraordinarily powerful in many ways, but because the newly introduced gene (transgene) adds to a chromosome at a random position, it leaves the resident copy of the gene intact. As a result, the transgene technique allows scientists to add genes; it does not permit them to subtract genes or to alter them at will.
Gene targeting
While Evans was tracking down embryonic stem cells, Mario Capecchi, at the University of Utah in Salt Lake City, and Oliver Smithies, first at the University of Wisconsin in Madison and later at the University of North Carolina in Chapel Hill, were trying to target genes by a process called homologous recombination — “homologous” because the incoming DNA sequence lines up with its twin target sequence in the chromosome and “recombination” because the incoming and target molecules break and rejoin with each other. Depending on the technical details, the process replaces one version of the gene with another or adds another copy of the gene in tandem. In 1982, Capecchi showed that mammalian cells contain efficient enzymatic machinery for mediating homologous recombination between DNA molecules injected into the cell. With the thought of exploiting this machinery to carry out homologous recombination between newly added DNA and the corresponding chromosomal sequence, Capecchi and Smithies had the bold idea that they could alter any gene in the cell by replacing it with a modified version. Many scientists doubted that this notion would pan out. How could a unique incoming gene successfully sift through the vast amount of DNA in mammalian chromosomes and correctly home to the proper site? But Capecchi and Smithies persevered.
Smithies devised a laborious — but extremely sensitive — method to find cells in which gene integration occurred at a chosen location. He aimed to detect such rare cells among the many more in which the gene would occur at random sites. The winning cell would contain a single piece of DNA that carried unique features of both the input gene and the resident gene — characteristics that had resided on two different molecules at the beginning of the experiment, but had become next-door neighbors through homologous recombination.
Smithies sequentially divided a population of 4400 cells into smaller and smaller pools, tracking the ones that contained pieces of DNA with diagnostic features of both the donor and recipient molecules. Eventually, he found a cell that carried the critical combination of DNA sequences on a single molecule, and published the work in 1985. His discovery showed that specific planned modification of native genes was possible. The DNA dart had found its corresponding sequence in the vast tangle of chromosomal DNA; he’d targeted a specific gene.
Capecchi took a different tack. First, his lab generated mammalian cells that contained insertions of a defective drug resistance gene. In a second step, the researchers introduced target DNA that carried a different defective version of the gene into the same cells. Homologous recombination between the two sequences could create an operational gene, and only cells in which the two defective versions recombined would resist the lethal effects of a toxic drug. This approach allowed scientists to easily find cells in which homologous recombination occurred, and helped them work out experimental conditions that made this process occur more efficiently.
These advances were big wins — but the game wasn’t over. How would scientists harness this capability to inactivate genes? And how would they extend the approach from cultured mammalian cells to the whole mouse?
Knocking out genes
Because the efficiency of gene targeting was low, the idea was to find the rare cultured ES cells that contain the new genetic alterations, and then implant those cells into early embryos. The resultant mice would breed to produce offspring that harbored the genetic change in every cell.
By 1987, both Capecchi and Smithies had shown that they could target a native gene in ES cells. They chose the same gene, hprt, for practical reasons. Because of the peculiarities of the gene’s function, the teams could isolate cells that contain defective or normal versions by exposing cells to different chemicals. This aspect of hprt allowed them to find knockout cells and also cells in which a mutation in the gene had been corrected. These experiments established the paradigm of knocking out a gene.
But most genes do not share these special properties of hprt. Capecchi wanted to develop a system that would be of general use in altering genes. Random recombination was 1000 times more frequent than homologous recombination so he needed to figure out a way not only to identify cells that carried the new DNA, but also to eradicate cells that carried it in the wrong site in the chromosome.
In 1988, Capecchi formulated a general strategy to enrich for cells in which the homologous targeting event had occurred. The scheme that he devised, now called the “positive-negative” method, not only enriches for recipient cells that have incorporated the DNA; it eliminates those that have allowed it to integrate at random sites. Most importantly, this technique makes it possible to replace virtually any gene of interest.
Making designer mice
In the meantime, Evans had been pursuing mouse embryonic stem cell technology — developing its use as a way to make designer mice. He and his student Allan Bradley and postdoctoral fellow Elizabeth Robertson had demonstrated that ES cells could generate chimeric animals capable of transmitting their genetic material to offspring. Furthermore, they had modified ES cells in culture dishes to create mice that would breed to produce baby mice with the genetic alterations in every cell.
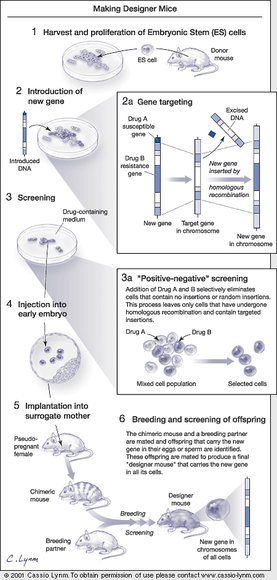
More than 4000 genetically engineered mice owe their existence to the technology developed by Capecchi, Evans, Smithies, and their colleagues—the procedure is pictured above in a simplified manner.
With this combination of Evans’s accomplishments in ES cell technology and Capecchi’s and Smithies’s achievements in directed gene targeting, one could now dream of altering any single gene in ES cells growing in culture, and then creating a mouse with the genetic change in all of its cells. In 1989, four groups realized this dream, each disabling a different gene.
A legacy
More than 4000 genetically engineered mice owe their existence to the technology developed by Capecchi, Evans, Smithies, and their colleagues. In addition to knocking out genes, the same method allows scientists to engineer precise changes in a particular gene. Insight into the processes to which these genes contribute is exploding as researchers assess the effects of the alterations, and pharmaceutical companies are using the resulting mouse models of disease to develop drugs. Refinements of the gene targeting technique are even allowing investigators to create a desired mutation in one particular tissue or at one particular time in the animal’s life. This ability allows scientists to study, for example, the effects of a gene in an adult mouse that is required for embryonic development. They can allow the animal to develop normally, and then inactivate the gene when desired.
Because genes influence nearly all biological phenomena, this technology is impacting the analysis in fields as diverse as cancer, immunology, neurobiology, human genetic disorders, endocrinology, and neurobiology. Capecchi, Evans, and Smithies have revolutionized the study of human health and disease.
by Evelyn Strauss
Key publications of Mario Capecchi
Capecchi, M.R. (1980). High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. 22, 479–488.
Folger, K.R., Wong, E.A., Wahl, G., and Capecchi, M.R. (1982). Patterns of integration of DNA microinjected into cultured mammalian cells: Evidence for homologous recombination between injected plasmid DNA molecules. Mol. Cell. Biol. 2, 1372–1387.
Thomas, K.R., Folger, K.R., and Capecchi, M.R. (1986). High frequency targeting of genes to specific sites in the mammalian genome. Cell. 44, 419-428.
Thomas, K.R. and Capecchi, M.R. (1987). Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 51, 503–512.
Mansour, S.L., Thomas, K.R., and Capecchi, M.R. (1988). Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: A general strategy for targeting mutations to nonselectable genes. Nature. 336, 348–352.
Capecchi, M.R. (1994). Targeted gene replacement. Sci. Am. 270, 54–61.
Capecchi, M.R. (1995). A personal view of gene targeting. Accomplishments in Cancer Research 1994, J.G. Fortner and J.E. Rhoads, ed,. Philadelphia: J.B. Lippincott, pp. 67–68.
Key publications of Oliver Smithies
Smithies, O. (1955). Zone electrophoresis in starch gels: group variations in the serum proteins of normal human adults. Biochem. J. 61, 629–641.
Smithies, O., Connell, G.E. and Dixon, G.H. (1962). Chromosomal rearrangements and the evolution of haptoglobin genes. Nature. 196, 232–236.
Slightom, J.L., Blechl, A.E., and Smithies, O. (1980). Human Fetal Gy- and Ay-globin genes: complete nucleotide sequences suggest that DNA can be exchanged between these duplicated genes. Cell. 21, 627–638.
Smithies, O., Gregg, R.G., Boggs, S.S., Koralewski, M.A., and Kucherlapati, R.S. (1985). Insertion of DNA sequences into the human chromosomal B-globin locus by homologous recombination. Nature. 317, 230–234.
Doetschman, T., Gregg, R.G., Maeda, N., Hooper, M.L., Melton, D.W., Thompson, S., and Smithies, O. (1987). Targeted correction of a mutant hprt gene in mouse embryonic stem cells. Nature. 330, 576–578.
Smithies, O., Kim, H.-S., Takahashi, N., and Edgell, M.H. On the importance of quantitative genetic variations in the etiology of hypertension. Kidney International (in press, 2000).
Award presentation by Ira Herskowitz
The release of the human genome sequence in draft form makes this a landmark year in the history of biology. Now we know that we have 30,000 or so genes (or is it 50,000?). We are now faced with several important questions, which include: First, what are the functions of these genes and the proteins that they code for? And, second, how can we use this information to improve human health?
Until the ability to knock out genes in the mouse was developed, determining the function of human genes seemed largely out of reach, tantalizingly so. For example, we might know of a human protein that is found only in certain cells of the brain and suspect what it might do, but how can we find out? Or, we might know of a gene in the fruit fly that is necessary for its development and see that humans have a very similar gene. Does it perform a similar function in humans? A powerful way to link a gene to function is to study the behavior of a mutant that lacks that gene and then see what the mutant can and cannot do. It’s somewhat like disabling an automobile by removing one part and then inferring the function of the part that was removed. But we can’t knock out genes in a human, so how can such mutants be produced?
The mighty mouse has come to the rescue. Its genes are typically 95 percent identical in sequence to ours, and we share the vast majority of our genes with the mouse.
Acceptance remarks
Mr. James W. Fordyce, members of the board of the Albert and Mary Lasker Foundation, Dr. Joseph Goldstein, members of the Awards Jury, and not least, all of the guests that are here with us to celebrate this special occasion, it is an enormous honor to have been selected as one of your recipients for the 2001 Lasker prize.
We are at the beginning of a new century, a new millennium and soon to reap many of the harvests accrued from the molecular biology revolution. It may not be too presumptuous to anticipate that this coming century will be noted by future historians as the Age of Biology. This revolution started on April 25, 1953.
I am a child, a product of the age of molecular biology. From its outset molecular biology was a new breed of science and scientists. Devotees from physics, chemistry and biology joined its ranks. Everything was new. There were no limitations. The guiding premise was that the most complex biological phenomena could, with persistence, be understood in molecular terms. Genetics, along with molecular biology, became the principal means for dissecting complex biological phenomena into workable subunits. Initially, the focus of molecular biologists was bacteria and bacterial viruses. Soon all organisms, including ourselves, came under their scrutiny.
I was first exposed to molecular biology at MIT. As an undergraduate at Antioch College, majoring in Chemistry and Physics, every alternate quarter we packed up our bags and set off for a new city to participate in work experience. I worked for several quarters in Dr. Alex Rich’s laboratory, at MIT. In addition to experimental work, I also had the opportunity to take courses from Salvador Luria, Boris Magasanick, and Cyrus Leventhal. They were not only great figures of that era, but tremendous teachers.
For my graduate studies, I decided to go to Harvard University and work for James Watson. Professor Watson had a profound influence on my career. He personified molecular biology. His bravado encouraged self-confidence in those around him. His stark honesty made our quest for truth uncompromising. He taught us not to bother with small questions, for such pursuits were likely to produce small answers.
Doing science in Jim’s laboratory was a blast. As a graduate student I was provided with what appeared to be limitless resources. I could not be kept out of the lab. We were cracking the genetic code, determining how proteins were synthesized and isolating the enzymatic machinery required for transcription. At this time, Walter Gilbert was also working in Jim’s laboratory. Jim and Wally complemented each other brilliantly because they had such different approaches to science. Jim was intuitive, Wally quantitative. As students, we received the benefits of both.
From Jim’s laboratory, I joined the faculty at Harvard Medical School and from there went to the University of Utah. When contemplating leaving Harvard and going to Utah, which most thought was quite rash, I asked Jim what he thought about the prospect of such a move. He looked at me and said that I could do good science anywhere. The move turned out to be a good decision. In Utah, I had the luxury to be able to pursue long-term projects, including gene targeting, that were not possible at Harvard, a bastion of short-term gratification.
I do not have time to go into a description of our contributions to gene targeting. For those who are interested in this history, I recommend to you articles soon to be published by Nature Medicine. Instead, I tried to recreate for you an image of an era that inspired and led to our experiments, and to give tribute to a few of the great figures that influenced my journey.
Though I was advised not to, I want to close by thanking three people in this audience. First, my mentor, Jim Watson, who taught me not so much how to do science, but the essence of science; Dr. Kirk Thomas, who has been a dear colleague in my laboratory for many years, and in particular during some of the crucial years when the development of the gene targeting technology occurred; and finally my wife, Laurie Fraser, who has had the patience to put up with me for so many years. I do some of my wildest thinking starting around 2:00 a.m. In my excitement, I nudge my wife. She awakens and says “What?” And I begin my story. By 5:00 a.m. I am satisfied that I have provided sufficient details to make the story compelling and then promptly fall to sleep. As I wander off into dreamland, I hear her whisper, “My God!”
Thank you for your patience. I wish to again express my gratitude to all of you for conferring this great honor upon me. Thank you.
Thank you so much…
In the light of the terrible events of the last weeks, I must take this opportunity to bring you a message of heartfelt sympathy and support from myself, my family, and many friends and colleagues. As a nation, we, too, have faced terrorism over many years, often externally supported. We are particularly heartened by your gritty, determined response to retain civilised normality — of which this ceremony is but one example. I am very pleased to be here today.
Thank you for having honored me in this manner. This is an awesome occasion — particularly important to me is the recognition which this award bestows upon my work.
The list of laureates represents a stellar collection of the greatest biomedical scientists, many of them my scientific heroes. I find myself both humbled and exhilarated in becoming one of their company.
I would not be here today if it were not for the love, belief and encouragement of my Father and Mother. Most important, too, has been the continued love and support of my wife Judith and of my family, my daughter Clare, who is here today, and my sons, Christopher and Simon, both of whom would also be here but for their heavy work commitments and young babies.
I must also mention just one scientific colleague — Matt Kaufman — who provided the final embryo trick which allowed me to realize my vision of establishing a pluripotential cell line directly from an embryo.
The words around the edge of this £2 coin say “Standing on the shoulders of Giants.” This motto is a reminder that progress of science is a cooperative as well as an individual activity. I have built upon earlier work and I am proud to enter the ranks of those who will help to shape the future by having provided a basis for so much further development.
Although experimental mammalian genetics with the use of ES cells and the techniques pioneered by my co-awardees is now well founded and used, there is still much to be learned and much interesting research in store about what genes really do in the context of the real biology of the whole organism in a complex environment.
I am particularly pleased that the fundamental knowledge of gene function and the derivation of animal models of human disease are underpinning important therapeutic advances.
There is another exciting development coming from ES cells focusing on their differentiative capacity. Human ES cells may become the basis for far-reaching progress in cellular therapy.
I have had a fascinating research career. I am pleased to see practical outcomes from fundamental investigation. This is a wonderful award and recognition.
Once again, many thanks.
An interview with Oliver Smithies by Raju Kucherlapati
Raju Kucherlapati, Scientific Director of the Harvard-Partners Center for Genetics and Genomics, interviews Oliver Smithies, whose work on homologous recombination led him to provide the groundwork to manipulate the mouse genome with exquisite precision.
Date of interview: September 20, 2001
Kucherlapati: I’m Raju Kucherlapati from the Harvard Medical School, and here with me today is Dr. Oliver Smithies. He’s the 2001 Albert Lasker Award winner for Basic Medical Research. Oliver, welcome.
Smithies: Thank you, Raju.
Kucherlapati: Oliver, first I want to congratulate you on this great accomplishment of being the Lasker Award winner this year.
Smithies: Thank you.
Kucherlapati: Oliver, it would be wonderful if you could tell everybody a little bit about your educational background.
Smithies: Yes, I would enjoy doing that because I have enjoyed my education. My primary education was of course in a little school in a country village near a big town. But then from that school I got to go to the local grammar school, as it was called in those days in England. A very distinguished school that was first opened in 1596, so it was a longstanding school and with a great scholastic tradition. And in that school when you got to the end of your time in it, you had the opportunity to be in the sixth form and to apply for scholarships.
So I applied for a scholarship under the guidance of my high school or grammar school teachers who were really marvelous people, and I got a scholarship to go to Oxford. Of all the colleges in Oxford — there are many colleges in Oxford, and you can apply for different colleges — I applied to go to Balliol, which is a college which was a very scholastic college. Now this was during World War II, so it was quite a small college then and you were taught by two levels. You were taught by the university professors and lecturers, and you were also taught by your tutor. He would be a very personal individual who you had personal contact with once a week.
They didn’t have very many students at that time, so the total college was only 90-odd students and my tutor probably had ten students. You would meet with him once a week and write an essay on a topic of his choice, not yours. This was a rather remarkable type of education because we were expected to read textbooks for one day of the week and then reviews for the second day, and then after that it had to be all original literature. So you got to enjoy learning from the original literature very early in your career and understanding the basic parts of science.
That was the undergraduate education. I took a degree in physiology, animal physiology, and I was actually a medical student. In actual fact, I dropped out of med-school and went to do chemistry, and then took a couple of years of chemistry and started to work with the same tutor now as my PhD advisor. And I did a PhD with him for two or three years, working on developing what we would now call a tool of biophysics. But that hadn’t been invented, there’s a name then and it was called “physical biochemistry” when I was doing it. So that was my primary education.
I should add that I lived in his house part of the time. We became very great friends; I felt part of the family. Because in World War II there weren’t enough places to live, so I stayed in their house and messed up their sink with my chemicals and got all of that by his wife, who was a marvelous person.
Anyway, then I went to the University of Wisconsin as a post-doc in physical chemistry and did a rather undistinguished couple of years of post-doc. Then, having been at a very susceptible age, I fell in love with an American woman — or girl I would have called her at that time — and we decided that it would be a good idea to get married. She didn’t want to go to England, so I went to Canada because of a visa problem.
I went to Canada and stayed in Canada for about seven years, and it was there where I did some work we might talk of later in relation to starch gel electrophoresis, and became a geneticist. I came back to University of Wisconsin at Madison again, but now as a geneticist on the faculty of genetics, and then became a professional geneticist. So that’s a rather long-winded account of what happened.
