
Douglas Coleman
Jackson Laboratory

Jeffrey M. Friedman
The Rockefeller University
For the discovery of leptin, a hormone that regulates appetite and body weight — a breakthrough that opened obesity research to molecular exploration.
The 2010 Albert Lasker Basic Medical Research Award honors two scientists for their discovery of leptin, a hormone that regulates appetite and body weight. Douglas Coleman (Jackson Laboratory) established that an appetite-suppressing substance circulates in the bloodstream and signals a second molecule to curb hunger. Jeffrey M. Friedman (Rockefeller University) isolated the gene that encodes the appetite suppressant and showed that fat cells release it. Their studies and subsequent findings demonstrated that this chemical, leptin, plays the central role in a self-regulating circuit: As fat accumulates, it exudes leptin, which binds to a receptor in the brain that quells the desire to eat. Coleman and Friedman have launched new understandings of obesity and disorders that result from perturbed leptin activity. They have overturned conventional notions with the insight that many overweight people suffer not from lack of willpower, but from metabolic disruptions.
Individual humans eat approximately a million calories per year, yet most people’s weight remains stable over long periods. By the 1950s, scientists were devising schemes for how the body balances food intake with energy expenditure. In 1953, Gordon C. Kennedy (National Institute for Medical Research, London) suggested that some compound, whose quantity mirrors that of stored fat, acts on the brain’s hypothalamus, a site that was known to house appetite centers. In the late 1950s, Romaine Hervey (University of Cambridge) created lesions in the hypothalamus that subsequently caused rats to eat voraciously. He then surgically connected pairs of animals — one with hypothalamic damage and one with an untouched brain — such that elements of their circulatory systems mixed. The normal rats stopped eating, Hervey reported. He proposed that chemical information from an overfed body travels through the bloodstream to intact appetite centers, which respond by subduing hunger in the normal animals.
Weighty connections
In 1965, Coleman began studying a mouse strain that had emerged at the Jackson Laboratory. Disturbances in its metabolism resembled those that occur in human diabetes, and one of its most striking characteristics was obesity. This mouse owed its troubles to two defective copies of a gene that researchers dubbed diabetes (db).
Coleman wondered whether something in the mutant mice might trigger problems in normal mice or, conversely, whether something in normal mice might cure the “diabetes” animals. He tested these ideas through an experimental approach similar to Hervey’s, in which scientists sew together two animals so that blood-borne factors can move from one individual to the other. If the sick mice harbored a harmful substance, it would enter the normal animals’ bodies and spur weight gain, Coleman reasoned. If the normal animals carried a health-promoting material, it would ease the sick animals’ symptoms.
The outcome proved more interesting than either of those possibilities, Coleman reported in 1969. Rather than gorging, as the “diabetes” mice did, the normal mice stopped eating and died, apparently from starvation. The ‘diabetes’ mice must have released a factor that quashed the normal animals’ drive to eat, Coleman surmised — a factor to which mutant animals could not respond.
To investigate further, he turned to another overweight mouse, this one called “obese,” whose aberrant physiology arises from two defective copies of a different gene (ob). Although the “obese” and “diabetes” mice displayed almost identical features — heft and other metabolic irregularities — they behaved radically differently after Coleman connected their circulatory systems. Nothing happened to the “diabetes” animals, but the “obese” mice starved to death, just as the normal rodents had in the previous experiment. In contrast, attaching normal mice to “obese” animals did not perturb the normal creatures and caused the “obese” ones to trim food consumption and gain less weight than usual.
Together, these results implied that normal mice make a substance that restrains appetite, but not to a dangerous degree. In contrast, “obese” mice do not manufacture an operational satiety factor, but they can detect and react to it. “Diabetes” mice overproduce the substance, thus causing normal and “obese” mice to stop eating — but they fail to respond to it. To validate these ideas, scientists needed to identify the db and ob genes and protein products, a task that posed insurmountable challenges at the time.
Hungry for genes
As a postdoctoral fellow, Jeffrey Friedman was captivated by how molecules control complex behaviors. His work led him to the “obese” mice and, by 1986, when he joined the faculty of Rockefeller University, he had decided to track down the ob gene. Researchers had just developed methods that allow scientists to isolate genes based on their location in the genome. Despite the technological power of this approach, the undertaking was enormously ambitious. Previous work had established that the ob gene resides within a large stretch of mouse chromosome 6, so Friedman, in collaboration with Rudolph Leibel (Rockefeller University), first narrowed its position to a much smaller region. Then Friedman and his lab members analyzed hundreds of molecular markers in almost 1000 mice to home in on it.
In 1994, Friedman’s team reported that it had isolated and sequenced the ob gene. The researchers then confirmed its identity by analyzing two strains of obese mice: One failed to manufacture ob messenger RNA, whereas the other overproduced a version of it that was predicted to encode a nonfunctional protein.
Normal animals displayed ob gene activity in fat, but not in other types of tissue, Friedman found, and he isolated the human version of the ob gene from fat cells. These observations were intriguing, given Kennedy’s proposal four decades earlier and the dogma that fat passively stores energy.
To explore the idea that fat produces an ob-encoded protein, which functions as a hormone, traveling elsewhere in the body to govern appetite, Friedman—and soon others—tested several predictions. First, the molecule should appear in the circulation. In 1995, Friedman’s group engineered bacteria to fabricate the protein (OB), generated antibodies that bind to it, and showed that all mammals tested, including humans and rodents, carry OB in their blood. Furthermore, “diabetes” mice make excess quantities of it, as predicted from Coleman’s experiments, and its amounts decrease in normal animals and obese humans after weight loss.
The model also implies that OB administration curtails food intake and body weight in “obese” and normal mice, but not in “diabetes” animals; Friedman and others confirmed this prediction. Furthermore, the protein exerts particularly potent effects when injected into the cerebrospinal fluid, suggesting that it acts directly on the brain. Because the ob-encoded protein makes animals slim down, Friedman named it leptin, from leptos, the Greek word for “thin.”
In late 1995, a group led by Louis A. Tartaglia (Millennium Pharmaceutical Incorporated, Cambridge, MA) identified a gene whose protein product binds leptin. Two months later, Friedman and Tartaglia independently showed that this leptin receptor is encoded by the diabetes gene and has multiple forms, one of which is defective in “diabetes” mice. Subsequent gene-activity studies from Friedman’s lab implied that the normal version of this receptor variant is especially abundant in the hypothalamus.
Together these results explained Coleman’s findings. Because the “diabetes” mice lack the receptor that senses the satiety factor, now known as leptin, hunger persists regardless how much food the animals consume. As they gain weight, the accumulating fat produces more and more leptin, which directs normal and “obese” mice to stop eating. “Obese” animals do not make functional leptin, but they respond to it when they receive it from “diabetes” or normal mice.
Fat chances
Friedman’s discoveries fueled intense efforts to uncover the mechanism by which leptin exerts its effects and apply these findings to clinical challenges. In 1997, Sadaf Farooqi and Stephen O’Rahilly (Addenbrooke’s Hospital, Cambridge) identified rare people with mutations in its gene and showed that they are extremely sensitive to leptin-replacement therapy (see photograph). Most obese individuals, however, possess large amounts of circulating leptin, yet remain overweight. Researchers are trying to understand why ordinary obese humans resist their own leptin and are strategizing how to counteract this situation.
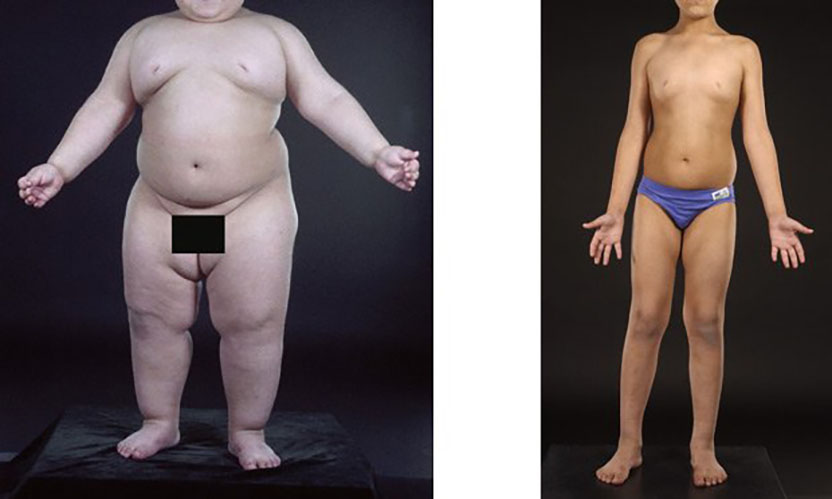
Enlightened treatment. Daily injections of leptin dramatically reduced appetite and weight in a child with leptin deficiency. At three years old (left), this boy was morbidly obese; at seven years old (right), he was in the 75th percentile for weight. (Courtesy of Stephen O’Rahilly and Sadaf Farooqi, with permission of the child’s parents.)
Scientists have also discovered that inadequate leptin underlies other pathologies. In lipodystrophy, for example, patients lack sufficient fat tissue and underproduce leptin; symptoms include insulin resistance and high cholesterol levels. Although inherited lipodystrophy is uncommon, a significant proportion of people with HIV acquire the condition. Preliminary studies suggest that leptin therapy can provide therapeutic benefits for these individuals. This treatment can also restore menstrual cycles that have stopped in women who subject themselves to extreme exercise regimes or reduced food intake.
Coleman and Friedman have fostered an explosion in our knowledge about how the body manages hunger and weight control. Leptin presides over a network that plays a crucial role in normal physiology and disease, and scientists have only begun to explore the myriad ways that we might manipulate this system to enhance people’s health and well being.
by Evelyn Strauss
Key publications of Douglas Coleman
Hummel, K.P., Dickie, M.M., and Coleman, D.L. (1966). Diabetes, a new mutation in the mouse. Science. 153, 1127-1128.
Coleman, D.L. and Hummel, K.P. (1967). Studies with the mutation, diabetes, in the mouse. Diabetologia. 3, 238-248.
Coleman, D.L. and Hummel, K.P. (1969). Effects of parabiosis of normal with genetically diabetic mice. Am. J. Physiol. 217, 1298-1304.
Coleman, D.L. and Hummel, K.P. (1970). The effects of hypothalamic lesions in genetically diabetic mice. Diabetologia. 6, 263-267.
Coleman, D.L. (1973). Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 9, 294-298.
Coleman, D.L. (1978). Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 14, 141-148.
Key publications of Jeffrey Friedman
Friedman, J.M., Leibel, R.L., and Bahary, N. (1991). Molecular mapping of obesity genes. Mammalian Genome. 1, 130-144.
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J.M. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature. 372, 425-432.
Halaas, J.L., Gajiwala, K.S., Maffei, M., Cohen, S.L., Chait, B.T., Rabinowitz, D., Lallone, R.L., Burley, S.K., and Friedman, J.M. (1995). Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 269, 543-546.
Lee, G.-H., Proenca, R., Montez, J.M., Carroll, K.M., Darvishzadeh, J.G., Lee, J.I., and Friedman, J.M. (1996). Abnormal splicing of the leptin receptor in diabetic mice. Nature. 379, 632-635.
DeFalco, J., Tomishima, M., Liu, H., Zho, C., Cai, X., Marth, J.D., Enquist, L., and Friedman, J.M. (2001). Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 291, 2608-2613.
Friedman, J.M. (2009). Leptin at 14 y of age: an ongoing story. Am. J. Clin. Nutr. 89, 973S-979S.
Award presentation by Michael Brown
 This year’s Lasker Basic Science Award honors two scientists who challenged our notion of free will. Let me explain. The thin among you take pride in your willpower. Through rigid self-control, you pass up rich deserts and you never touch a French-fried potato or a pepperoni pizza. Only the slothful succumb. Today’s honorees taught us that you shouldn’t be so smug. Your willpower alone doesn’t protect you from gluttony. It needs help from your leptin.
This year’s Lasker Basic Science Award honors two scientists who challenged our notion of free will. Let me explain. The thin among you take pride in your willpower. Through rigid self-control, you pass up rich deserts and you never touch a French-fried potato or a pepperoni pizza. Only the slothful succumb. Today’s honorees taught us that you shouldn’t be so smug. Your willpower alone doesn’t protect you from gluttony. It needs help from your leptin.
Let me illustrate. Each of you received a booklet describing today’s ceremonies. Please turn to page 15. At the top you see a three-year-old baby who weighs 92 pounds. Imagine hefting a 92-pound baby around the zoo. The baby is fat because he never stops eating. His body cannot produce a hormone called leptin. His two genetic templates for leptin are both defective. Leptin is a hormone that circulates in the blood and enters the brain, where it makes us stop eating. If our bodies lack leptin, we are condemned to eat ourselves to death, willpower or no.
Now look at the picture on the bottom. This is the same boy at age 7. His parents have injected him daily with leptin. His appetite is tamed and he has joined the ranks of the strong-willed. Not because of his will, but because of his leptin. The lesson is clear. If any of you were deficient in leptin you would weigh 400 pounds. Right now you would be begging for a second helping of desert. So much for free will.
Acceptance remarks
Acceptance remarks, 2010 Lasker Awards Ceremony
I am overwhelmed and humbled to be selected as a co-winner of the 2010 Lasker Basic Medical Research Award. I have always viewed the Lasker as one of the most esteemed biomedical awards, and it is with great pride that I accept this honor.
As the only son of parents who had little formal education, I was encouraged to excel in school. During undergraduate studies at McMaster University, a very dynamic professor taught me the rudiments of biochemistry and an appreciation of the scientific method. With his enthusiastic support, I entered graduate school at the University of Wisconsin and received my PhD in 1958. Rather than starting postdoctoral studies or a career in academia or industry, I became a research scientist at The Jackson Laboratory. This decision was transformative: The Jackson Laboratory had fascinating genetic disease models, interactive colleagues, and Acadia National Park as a backyard.
Initially, I had no intention of studying the diabetes/obesity syndrome, but in 1965 a spontaneous mouse mutation was discovered and I began research that would consume much of my scientific thought for the better part of three decades. This new mutant was similar to a previously characterized obese mutant, except that it displayed severe life-shortening diabetes. Operating on the hypothesis that a blood-borne factor might regulate the severity of diabetes in these mutants, I used a technique called parabiosis to mix and match mutant and wild-type blood supplies. Based on these experiments, I concluded that the diabetes mutant overproduced, but did not respond to, a blood-borne ‘satiety factor’, while the obese mutant recognized this factor but was unable to produce it. Subsequent studies suggested that adipocytes synthesized this factor and the hypothalamus contained its receptor. The scientific community did not readily accept these conclusions because obesity was considered strictly a behavioral problem, not a physiological problem.
Definitive proof of my conclusions required isolating the satiety factor — a feat that resisted rigorous experimentation. Finally, in 1994 Jeffrey Friedman succeeded in identifying the obese gene and demonstrated that it encoded the powerful hormone leptin. With the subsequent cloning of the leptin receptor, the field exploded. Essentially all of my predictions were verified: the obese mutant lacks active leptin, which is normally produced in adipose tissue; the diabetes mutant lacks an active leptin receptor, which is normally expressed in the hypothalamus. With these findings, two long-standing misconceptions were definitively laid to rest: obesity was not merely a behavioral problem but rather had a significant physiological component, and adipose tissue was not merely a fat-storage site but rather an important endocrine organ.
In closing, special thanks go to my family who were not only supportive of my work but a welcome diversion from it. My wife for nearly 55 years, Beverly, merits my strongest gratitude because she, more than anyone, built the love, support, and understanding that was the foundation on which I functioned. She would have been delighted to share in the accolades from this most prestigious award.

Acceptance remarks, 2010 Lasker Awards Ceremony
It is a profound honor to receive this year’s Lasker Award for Basic Research. The significance of this prize is heightened by the ground-breaking achievements of the prior recipients, who are among the greatest contributors to medical science, and the depth of the esteem in which each of the committee members is held. I would also like to thank the many people who have helped me, and a list of these people’s names are included in my full acknowledgements in the Lasker brochure.
In 1903, Theodore Roosevelt wrote, “Far and away the best prize that life offers is the chance to work hard at work worth doing.” While there is wisdom in this statement, I have come to conclude that it fails to capture that which motivates me. For me the best prize was having had the chance to make a discovery.
As scientists we are the instruments of a wave of new knowledge that passes through of us from the past and into the future. The wave of knowledge of which my research is a part began with the realization of the French chemist Lavoisier in the 18th century that living organisms are subject to the basic laws of physics and chemistry, the formulation of the first law of thermodynamics by Joule and Meyer, passes through Hetherington and Ranson, two great neuro-anatomists, and Gordon Kennedy and Romaine Hervey, two physiologists of the 20th century who suggested that fat-tissue-generated signals that regulated body weight. In a beautiful set of experiments for which he is being recognized, Doug Coleman of The Jackson Laboratory correctly predicted that this factor was encoded by the mouse ob gene.
This wave of new knowledge coursed briefly through me, highlighted by a singular moment when the presence and absence of a few regions of intensity on an X-ray revealed the answer to a simple question that had perplexed scientists since Lavoisier: How do biologic systems count calories? Or more precisely, how does Nature monitor the number of calories stored by fat? We now know that this is achieved by the production of a hormone by fat tissue, and it was the realization that the amount of body fat is regulated by this hormone, leptin, that led me to be recognized today. This moment of discovery was exhilarating. Gazing upon the X-ray was also humbling because it revealed the beauty and power and majesty of Nature, which had solved the problem of counting calories so elegantly and simply. The only moments in my life that compared to this were when I was married and when I heard my twin daughters cry for the first time as they were delivered.
This wave of knowledge that began with Laviosier and passed through Doug, then me, will continue into a future that I cannot clearly see but which others undoubtedly can or will. This future puts forth the promise that in time we will understand how complex behaviors such as feeding are regulated at the cellular and the molecular level.
The realization that leptin and other molecules control feeding behavior and body weight show that obesity is not a personal choice. It is my hope and expectation that the realization that obesity has a biologic basis will not only lead to new treatments but also lead to a greater sense of understanding for the obese. It is simplistic to imagine that we can consciously control all of our basic drives, drives that have been honed by evolution for countless millennia.
As a child, it would have been inconceivable to me that I would ever have my name listed with the previous recipients of this award, and it is the deepest of honors. The depth of this occasion is completed by the presence of my friends, colleagues, and family, in particular my wife, Lily, and my daughters, Nathalie and Alexandra.
Interview with Doug Coleman and Jeffrey Friedman
Video Credit: Susan Hadary
