
William G. Kaelin, Jr.
Dana-Farber Cancer Institute, Harvard Medical School

Peter J. Ratcliffe
University of Oxford, Francis Crick Institute

Gregg L. Semenza
Johns Hopkins University School of Medicine
The 2016 Albert Lasker Basic Medical Research Award honors three physician-scientists for their discovery of the pathway by which cells from human and most animals sense and adapt to changes in oxygen availability, a process that is essential for survival. Scientists had long appreciated that the success of today’s dominant life forms hinges on oxygen, yet little was known about their responses to it. William G. Kaelin, Jr. (Dana-Farber Cancer Institute/Harvard Medical School), Peter J. Ratcliffe (University of Oxford/Francis Crick Institute), and Gregg L. Semenza (Johns Hopkins University School of Medicine) illuminated the core molecular events that explain how almost all multicellular animals tune their physiology to cope with varying quantities of the life-sustaining element, thus exposing a unique signaling scheme.
Energizing hypoxic genes
Animals require oxygen to extract energy from food, but too much of the chemical creates peril, as certain oxygen-containing compounds wreak molecular havoc. To handle this challenge, organisms have evolved honed systems to furnish optimal supplies. For example, many creatures use red blood cells to transport oxygen to tissues deep within their bodies. When its concentration sags, the body generates the hormone erythropoietin, which boosts red blood-cell production.
In the early 1990s, Semenza and Ratcliffe were trying to understand how oxygen deprivation—hypoxia—triggers the erythropoietin gene. They and others identified a DNA sequence that is necessary for hypoxia-dependent activation. Placement of this DNA stretch next to other genes renders those genes inducible by low-oxygen conditions too. Semenza showed that protein from the nucleus sticks to this DNA-control region, but only when oxygen is scarce. Furthermore, sequence alterations that eliminate protein binding to DNA also obliterate hypoxia-spurred gene stimulation. Perhaps, he proposed, the nuclear protein, which he called Hypoxia-Inducible Factor 1 (HIF-1), sits down on DNA when cells lack oxygen and rouses adjacent genes.
Semenza and postdoctoral fellow Guang Wang achieved a major breakthrough in 1995, when they purified HIF-1 and found that it contains two protein partners, HIF-1α and HIF-1β. The HIF-1α component was novel, and they isolated its gene from human cells. HIF-1α vanished quickly when Semenza shifted cells from low- to high-oxygen conditions.
A profusion of HIF-1-driven genes
Erythropoietin in adults is produced predominantly by specialized kidney cells, and scientists had long thought that mechanisms governing its manufacture were peculiar to those cells. With HIF-1 and its binding sequences in hand, Ratcliffe and Semenza upended this idea.
In complementary work, the scientists showed that hypoxia provokes HIF-1 to bind and activate target genes in many mammalian cells that do not make erythropoietin. Their observations suggested that multiple kinds of cells use HIF-1 to goad numerous genes in response to low-oxygen conditions.
Semenza and Ratcliffe subsequently confirmed this idea, and the list of hypoxia-induced genes expanded rapidly. In 1996, for instance, Semenza demonstrated that HIF-1 activates the gene for a key participant in blood-vessel formation, vascular endothelial growth factor (VEGF). This result extended HIF-1 into another key system by which the body can augment oxygen delivery. These and other findings established that HIF-1 lies at the core of an elaborate physiological network that ensures advantageous responses to oxygen.
Smothering HIF-1
The observation that the amount of HIF-1 plummets when cells are shifted to high-oxygen conditions squared with the factor’s hallmark ability to activate target genes only when oxygen is limited. It also raised a crucial question: What drives HIF-1 destruction? The answer came from an unexpected direction.
A familial cancer syndrome called von Hippel-Lindau (VHL) disease owes its pathology to faulty versions of a particular protein. William Kaelin was trying to figure out how defects in this VHL protein cause the illness. The classic VHL tumor is composed of inappropriate, newly formed blood vessels, and surplus VEGF characterizes these growths; excessive erythropoietin production also can occur. Kaelin wondered whether VHL influences activity of the genes for these and other hypoxic-induced substances.
In 1996, he and his colleagues grew human cells with and without operational VHL. Then they measured the abundance of several messenger RNAs, including VEGF’s, that normally disappear in response to oxygen. Even when oxygen was plentiful, VHL-defective cells contained large amounts of these mRNAs. Addition of intact VHL restored normal hypoxia-dependent quantities.
Kaelin then showed that VHL’s capacity to quash the accumulation of particular mRNAs in rich oxygen conditions relies on its ability to physically assemble with several other proteins, including one that was later shown to mark certain proteins for destruction by attaching the chemical tag ubiquitin. Ubiquitin is the signal that sends certain proteins to the cell’s incinerator, the proteasome (Albert Lasker Basic Medical Research Award, 2000). Research soon established that HIF-1α is degraded through the ubiquitin pathway when oxygen is profuse.
In 1999, Ratcliffe and colleagues demonstrated that the oxygen-dependent elimination of HIF-1α depends on VHL. Furthermore, under conditions that jam the proteasome, VHL and HIF-1α are present in the same protein assembly. Ratcliffe proposed that VHL interacts with HIF-1α under high-oxygen conditions and targets it for destruction.
Kaelin subsequently found that VHL binds directly to HIF-1α—and that optimal binding requires a region in HIF-1α that was known to be needed for its oxygen-triggered destruction. He, Ratcliffe, and others showed that the same region is ubiquitinated and that defects in VHL prevent addition of the chemical tag. Together, these observations established that the VHL assembly directs ubiquitination of HIF-1α when oxygen is present.
A crucial oxygen-based modification
Ratcliffe and Kaelin wondered what allows VHL to bind HIF-1α under high- but not low-oxygen conditions. Independently, they used various tricks to block the ubiquitin pathway, thereby allowing HIF-1α to accumulate even when oxygen abounds. Their observations suggested the existence of an enzyme that modifies HIF-1α such that it can snag VHL.
To identify the presumptive HIF-1α modification, both teams homed in on the region of HIF-1α that grabs VHL. Elimination of a single amino acid—a proline—in this region abrogates VHL’s ability to attach ubiquitins and thus safeguards HIF-1. Further analysis revealed that this proline had picked up an oxygen atom next to one of its hydrogens. HIF-1α had thus acquired a chemical modification called a hydroxyl group.
Together, these and other findings indicated that a prolyl hydroxylase, named for its deeds, adds a hydroxyl to a critical proline in HIF-1α, thereby rendering HIF-1α recognizable by VHL and fostering its consequent ruin. Because prolyl hydroxylases require molecular oxygen to do their jobs, the observations, published in 2001, explained why HIF-1α is not degraded under hypoxic conditions and thus, how the enzyme translates oxygen levels into HIF-1α stability (see figure).
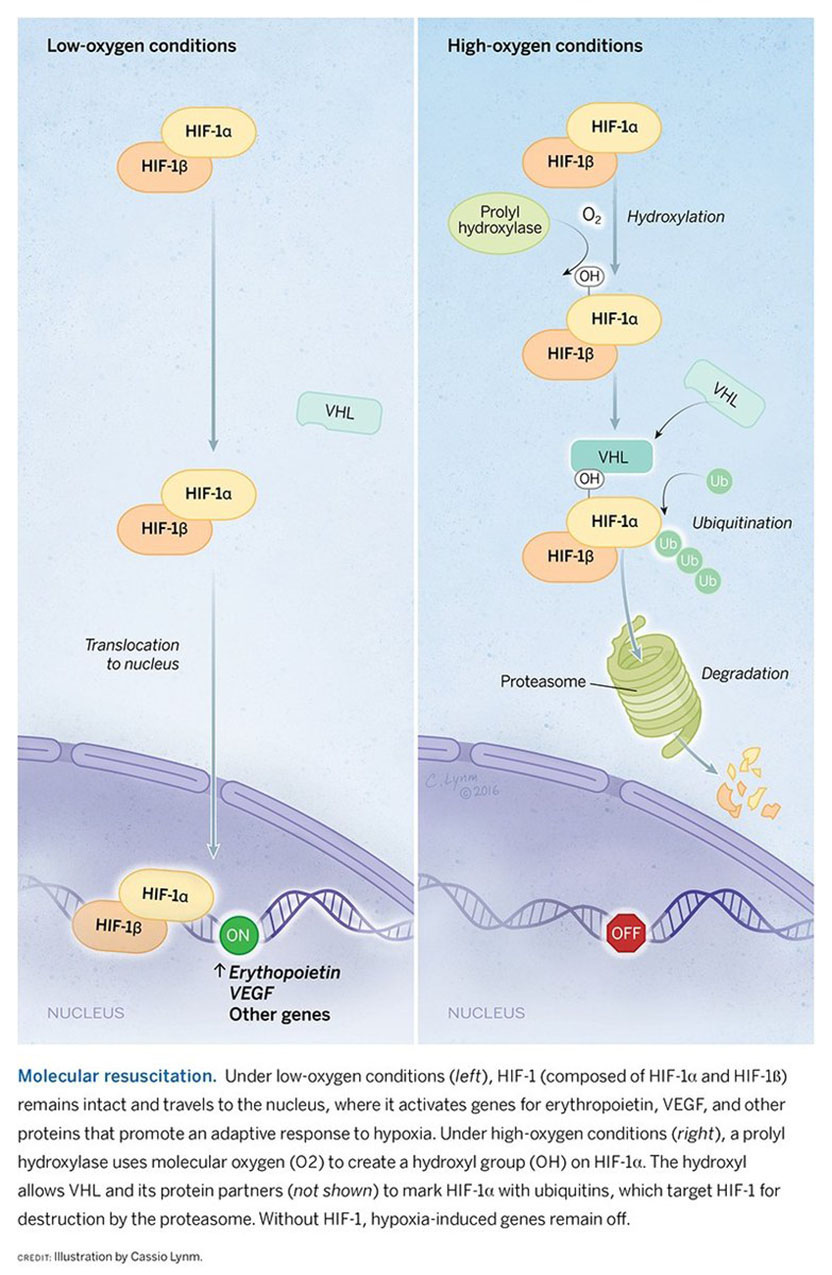
The experiments had not, however, uncovered the specific prolyl hydroxylase that acts on HIF-1α, and the amino acid sequence around the hydroxylated HIF-1α proline did not match target sites for known prolyl hydroxylases. In collaboration with Christopher Schofield (University of Oxford), Ratcliffe proposed that the enzyme belongs to a larger molecular family. Ratcliffe and, independently, Steven McKnight (University of Texas Southwestern Medical Center) identified three related prolyl hydroxylases that govern the cellular response to oxygen in mammals.
Ratcliffe subsequently demonstrated that hydroxylation of another proline in HIF-1α also promotes its oxygen-dependent, VHL-mediated demolition. Soon afterward, others found that hydroxylation of a different amino acid incapacitates HIF-1α as well, but in a different way.
Breathing new life into therapies
We now know that HIF-1 and its molecular relatives regulate a plethora of genes whose products influence a vast number of biological processes. Harnessing the HIF pathway for therapeutic purposes hence offers fertile potential.
For example, prolyl hydroxylase inhibitors that preserve HIF and thus incite erythropoietin gene activity hold hope of reversing anemia. Results in animals as well as humans show promise, and several companies are pursuing this possibility. Interfering with HIF prolyl hydroxylases might also promote blood-vessel growth and other adaptations to hypoxia that would combat conditions resulting from inadequate circulation.
On the flip side, thwarting HIF might quell malignancies, in part because many tumors draw nourishment from extra blood vessels they construct. A compound that foils HIF-2α, a close relative of HIF-1α, is currently in early clinical trials for kidney cancer.
Kaelin, Ratcliffe, and Semenza have unveiled the fundamental workings of HIF, which presides over a complex and exquisitely controlled response to oxygen fluctuations. Their discoveries have revealed secrets about a cornerstone of life on Earth.
by Evelyn Strauss
Key Publications of William G. Kaelin, Jr.
Iliopoulos, O., Levy, A.P., Jiang, C., Kaelin, W.G., Jr., and Goldberg, M.A. (1996). Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA. 93, 10595-10599.
Ohh, M., Park, C.W., Ivan, M., Hoffman, M.A., Kim, T.-Y., Huang, L.E., Chau, V., Pavletich, N., and Kaelin, W.G., Jr. (2000). Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2, 423-427.
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J.M., Lane, W.S., and Kaelin, W.G., Jr. (2001). HIF targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 292, 464-468.
Ivan, M., Haberberger, T., Gervasi, D., Michelson, K.S., Gunzler, V., Kondo, K., Yang, H., Sorokina, I., Conaway, R.C., Conaway, J.W., and Kaelin, W.G., Jr. (2002). Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA. 99, 13459-13464.
Kaelin, W.G., Jr. and Ratcliffe, P.J. (2008). Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 4, 393-402.
Key Publications of Peter J. Ratcliffe
Maxwell, P.H., Pugh, C.W., and Ratcliffe, P.J. (1993). Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism. Proc. Natl. Acad. Sci. USA. 90, 2423-2427.
Maxwell, P.H., Wiesener, M.S., Chang, G.-W., Clifford, S.C., Vaux, E.C., Cockman, M.E., Wykoff, C.C., Pugh, C.W., Maher, E.R., and Ratcliffe, P.J. (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 399, 271-275.
Jaakkola, P., Mole, D.R., Tian, Y.-M., Wilson, M.I., Gielbert, J., Gaskell, S.J., von Kriegsheim, A., Hebestreit, H.F., Mukherji, M., Schofield, C.J., Maxwell, P.H., Pugh, C.W., and Ratcliffe, P.J. (2001). Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 292, 468-472.
Epstein, A.C.R., Gleadle, J.M., McNeill, L.A., Hewitson, K.S., O’Rourke, J.F., Mole, D.R., Mukherji, M., Metzen, E., Wilson, M.I., Dhanda, A., Tian, Y.-M., Masson, N., Hamilton, D.L., Jaakkola, P., Barstead, R., Hodgkin, J., Maxwell, P.H., Pugh, C.W., Schofield, C.J., and Ratcliffe, P.J. (2001). C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 107, 43-54.
Cockman, M.E., Lancaster, D.E., Stolze, I.P., Hewitson, K.S., McDonough, M.A., Coleman, M.L., Coles, C.H., Yu, X., Hay, R.T., Ley, S.C., Pugh, C.W., Oldham, N.J., Masson, N., Schofield, C.J., and Ratcliffe, P.J. (2006). Post-translational hydroxylation of ankyrin repeats in IkB proteins by the HIF asparaginyl hydroxylases, FIH. Proc. Natl. Acad. Sci. USA 103, 14767-14722.
Key Publications of Gregg L. Semenza
Semenza, G.L., Nejfelt, M.K., Chi, S.M., and Antonarakis, S.E. (1991). Hypoxia-inducible nuclear factors bind to an enhancer located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA. 88, 5680-5684.
Semenza, G.L. and Wang, G.L. (1992). A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 12, 5447-5454.
Wang, G.L. and Semenza, G.L. (1995). Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 270, 1230-1237.
Wang, G.L., Jiang, B.-H., Rue, E.A., and Semenza, G. L. (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA. 92, 5510-5514.
Forsythe, J.A., Jiang, B.-H., Iyer, N.V., Agani, F., Leung, S.W., Koos, R.D., and Semenza, G.L. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604-4613.
Acceptance remarks
Acceptance remarks, 2016 Lasker Awards Ceremony
I grew up during the space race, so science and engineering were celebrated and supported throughout my childhood and our household had toys that fostered curiosity and creativity, including a microscope and chemistry sets.
In high school, I attended an NSF-sponsored summer program for 32 mathematically gifted students that changed my life. I was delighted to learn that I wasn’t the least gifted of the 32 but I certainly had, due to my abysmal study habits, the lowest grades. I learned that school was more fun when I was challenged, that it is really helpful to be surrounded by people who are smarter than you are, and that I, too, could get good grades if I actually did my homework.
As a premed, I floundered in a research laboratory working on an independent study project that was uninteresting, unimportant, and, as I would later correctly show, undoable. My mentor rewarded me with a “C+,” noting on my transcript that “Mr. Kaelin appears to be a bright young man whose future lies outside of the laboratory.” This painful experience convinced me I was going to be a clinician rather than a scientist, and I approached my clinical training accordingly. I even served as Chief Resident at Johns Hopkins, honing my knowledge of obscure diseases such as von Hippel-Lindau Disease.
Years later, I discovered, thanks to outstanding mentorship from David Livingston, that I actually could do science. And my clinical practice convinced me that we desperately needed better cancer treatments based on a deeper understanding of cancer pathogenesis.
I like mathematics, medicine, and science because I like solving puzzles, and I like answers that are objectively verifiable. Science gathers knowledge, and engineering applies that knowledge to useful purposes. For most of my career, I benefited from the wise decision in our country to have the public sector support basic scientific research, where the timelines and deliverables are too unpredictable for investors, and to let the private sector decide when a line of investigation is ripe for application and commercialization. Although this bargain served American biomedical research well, one hears repeated calls to treat science as though it were engineering, tying funding to anticipated outcomes and impact. This is troubling because many practices that are useful for managing engineering projects are antithetical to good science. For example, building teams and harmonizing goals is often essential for large engineering projects. Early stage science, however, is often driven by creative individuals who follow their curiosity and are willing to go where their science takes them. Forcing scientists into teams and holding them to predetermined deliverables can create herd mentality and stifle the heretical thinking that is often needed for transformative discoveries.
JFK knew it would take a decade to put a man on the moon because it was fundamentally an engineering challenge rather than a scientific challenge. It does scientists and, more importantly, patients and their families, a disservice when fundraisers and policy makers overpromise and oversimplify with respect to our greatest biomedical challenges, including cancer.
I am very proud to receive this award on behalf of the talented young scientists who have worked in my laboratory over the years, to share it with such esteemed colleagues, and to dedicate it to my beloved wife Carolyn, who died last summer.
Acceptance remarks, 2016 Lasker Awards Ceremony
I am deeply honoured to receive the Lasker Foundation Award for Basic Medical Research today.
I’d like to reflect for a moment on the many twists and turns that brought me to this fortunate position. One still clear in my mind dates to the Lancaster Royal Grammar School, circa 1970. I was a tolerable schoolboy chemist and intent on a career in industrial chemistry. The ethereal (but formidable) Headmaster appeared one morning in the chemistry classroom. ‘Peter’ he said with unnerving serenity ‘I think you should study medicine’. And without further thought, my university application forms were changed. To this day, I am unsure whether he felt I would be a good doctor or a bad chemist. But the experience is (I think) a reminder of the role of serendipity in a scientific career, at least in mine.
I did train in medicine, as a nephrologist, and came to research rather late, fascinated by the extraordinary sensitivity with which the kidneys regulate the hormone erythropoietin in accordance with blood oxygen content. I felt that the process was interesting and might be tractable. At the time, the erythropoietin gene had recently been identified, so there was a new opportunity for study. But some felt, with the emerging success of recombinant erythropoietin treatment, that understanding how the hormone was regulated was niche area, unlikely to be of general importance.
Acceptance remarks, 2016 Lasker Awards Ceremony
Many of us who conduct biomedical research do so with what could be described as a religious fervor. This would not have come as a surprise to Mary Lasker. She once told a reporter, “I am opposed to heart attacks and cancer the way one is opposed to sin.”
Amen.
Seven months ago, Antonin Scalia died. He had a heart attack, which occurs when the flow of blood through one of the coronary arteries is blocked, cutting off the heart muscle’s supply of oxygen.
Eight months ago, David Bowie died of liver cancer. Cancer cells invade surrounding tissue, make their way into blood vessels, and spread throughout the body. What are they searching for? My guess is oxygen.
Over the last 25 years, we’ve worked to understand how a family of proteins, which we called hypoxia-inducible factors, controls the ability of cells, tissues, and organ systems to respond to changes in oxygen availability. Our work began with an attempt to figure out how expression of the erythropoietin gene was turned on when the body was deprived of oxygen. Now we know that more than 2,000 other genes are regulated in a similar manner.
Looking forward, I’d like to think that within the next decade drugs that stimulate hypoxia-inducible factors will be used to treat anemia and cardiovascular disease, while drugs that inhibit these factors will prolong the lives of cancer patients.
So this is my religion: I am filled with Wonder at the outcome of 4 billion years of evolution here on our speck in the universe and Hope regarding our opportunity to improve the lives of those around us through basic science discoveries and their translation to clinical practice.
Well, I began with Mary Lasker, so I’ll end with Albert. As you know, Mr. Lasker was a successful advertising executive. He was in charge of accounts for products like Sunkist oranges and Pepsodent toothpaste before he and Mary began their crusade to promote biomedical research. Coincidentally, my grandfather also worked for an advertising agency here in Manhattan. His account was for a product called Wonder Bread.
Thank you.
The 2016 Basic Award video
Video Credit: Flora Lichtman
