
James P. Allison
University of Texas MD Anderson Cancer Center
For the discovery and development of a monoclonal antibody therapy that unleashes the immune system to combat cancer.
The 2015 Lasker~DeBakey Clinical Medical Research Award honors a scientist who discovered and developed a monoclonal antibody therapy that unleashes the immune system to combat cancer. By blocking a protein that normally restrains the body’s natural ability to attack tumor cells, James P. Allison (University of Texas MD Anderson Cancer Center) devised a fundamentally new strategy for treating malignancies. Because this approach targets immune cells rather than specific tumors, it holds great promise to thwart diverse cancers.
Allison’s work has already benefited thousands of people with advanced melanoma, a disease that typically used to kill people in less than a year. The therapy he conceived has delivered recoveries that last for a decade or more.
Turning on T cells
As the T cells of the immune system develop during neonatal life, they are trained to detect aberrant cells. After they complete this schooling, they are ready to scour the body for microbial interlopers and signs of cellular misbehavior, such as malignancy. When they perceive such hazards, they proliferate and recruit other elements of the immune system into service.
To identify trouble, T cells use a protein on their surfaces called the T cell receptor (TCR). The TCR binds to foreign bits of protein that are displayed on other cells. When presented in a particular way, the stimulant proteins spur the TCR to provoke T cells, which in turn spark an immune response that kills infected or otherwise problematic cells. In the early 1990s, researchers who study cancer were trying to exploit this phenomenon by goading the immune system with tumor proteins, but this vaccine approach wasn’t working.
By then, scientists had realized that naïve T cells — those that have not yet encountered foreign protein fragments — need more than engagement between the TCR and the presented proteins to jump into action. Experiments suggested that a T-cell protein called CD28 could supply the required extra jolt, and Allison established that it indeed does. Simultaneous stimulation by CD28 and the TCR spurs naïve T cells to proliferate and maintains their reactivity.
Previous sequence analysis of CD28 revealed that it resembles another protein, CTLA-4, which appears only on activated T cells. Furthermore, CD28 and CTLA-4 bind the same two proteins on cells that present proteins to T cells. Given the importance of CD28, immunologists were keen to understand its relative, CTLA-4.
Activating controversy
Studies from several groups in the early 1990s led to contradictory ideas about CTLA-4’s function. Initial findings suggested that it augments T-cell activation, but Allison and, independently, Jeffrey Bluestone (University of California, San Francisco) in collaboration with Craig Thompson (Memorial Sloan-Kettering Cancer Center) realized that providing a stimulator looks the same experimentally as removing a blockade. The researchers devised ways to distinguish between these possibilities.
In 1994 and 1995, respectively, Bluestone and Allison showed that CTLA-4 subdues T-cell activation. A picture began to emerge in which CTLA-4 and CD28 deliver opposing signals that the T cell integrates. In this view, CD28 enflames T cells and then CTLA-4 production kicks in and reins in a response that might otherwise cause overzealous reactions and autoimmune trouble.
Not everyone was convinced, and controversy continued to rage until two groups engineered mice that lack CTLA-4. These animals’ organs fill up with T cells and they die at a few weeks of age. This observation established that CTLA-4 inhibits T-cell proliferation, and it gave Allison an idea.
Perhaps attempts at creating anti-cancer vaccines failed because researchers were inadvertently pulling the CTLA-4 immune-response brake. The same proteins on cells that present foreign protein, after all, prod CTLA-4 as well as CD28. In this view, the immune system is poised to wipe out cancer cells, but CTLA-4 holds it in check. Perhaps by blocking CTLA-4–generated signals, Allison could liberate it to attack malignant cells.
To test this hypothesis, he and postdoctoral fellow Dana Leach developed an anti-CTLA-4 antibody and injected it into tumor-bearing mice. The animals rejected their tumors. In contrast, the cancerous growths progressively expanded for several weeks in untreated animals or those that received anti-CD28.
One of the hallmarks of the immune system is that it ‘remembers’ what it has encountered in the past. Upon spotting trespassers for the second time, it responds more quickly and forcefully than it did initially. Allison found that the animals fought off tumor cells that were injected more than two months after the original experiment. These and other results indicated that tumor rejection achieved by blocking CTLA-4 creates immunological memory.
Finally, Allison demonstrated that anti-CTLA-4 therapy could sustainably battle established tumors. He treated the mice after they had palpable masses, and this procedure worked even better than when the antibody was administered simultaneously with tumor injection.
Together, these results and others suggested that foiling CTLA-4 inhibitory activities held tremendous therapeutic promise. The approach diverged dramatically from existing strategies, which targeted cancer cells. In Allison’s vision, a single drug would unleash the immune system to quell any malignant threat, harnessing the body’s natural capacity to eliminate miscreant cells.
If it worked, this treatment also would elude resistance, a common plague of chemotherapy. As tumors displayed different proteins over the course of treatment, immune cells would tackle their ever-morphing foe, geared as they are to recognize an endless variety of intruders.
Struggling to bring anti-CTLA-4 to market
To deliver these hopes to the clinic, Allison needed a corporate partner — an outfit that would develop a therapeutic antibody for human use. He talked to many companies, but decision-makers were skittish about backing such a radical approach. The notion that a drug could fight cancer without targeting a specific tumor challenged conventional thought, as did the idea that an agent could exploit the immune system without directly stimulating it. Furthermore, the serious autoimmune effects in mice that lack CTLA-4 gave people pause. Allison continued to push for someone to pick up the project.
Finally, he snagged the interest of Alan Korman, a colleague and old friend. Korman joined forces with Nils Lonberg at a biotechnology company called Medarex, where scientists had developed a way to make human antibodies in mice. By 2000, Korman and Lonberg had used this approach to create a human antibody, ipilimumab, that binds and inactivates CTLA-4.
Medarex launched small clinical trials on various types of cancer, including metastatic melanoma. Responses looked somewhat promising, but worrisome side effects emerged. Furthermore, it became clear that measures for therapeutic success that had been developed for chemotherapeutic drugs were inappropriate for immunotherapies. Conventional criteria emphasized speed of response and tumor shrinkage, which made sense for agents that directly kill tumors. Anti-CTLA-4, however, built the immune response, which took time; tumors did not wither immediately. In some cases, they even appeared to grow initially, as immune cells flocked to the tumors.
Jedd Wolchok (Memorial Sloan-Kettering Cancer Center) and F. Stephen Hodi (Dana-Farber Cancer Institute,), in conjunction with scientists at Bristol-Myers Squibb Company, which had acquired Medarex, spearheaded an effort to devise new guidelines to assess patient benefit from immune therapies. They also developed ways to handle treatment-associated toxicities.
In 2010, Hodi and colleagues reported results from the first phase III randomized trial of ipilimumab in individuals with metastatic melanoma. Anti-CTLA-4 improves overall survival — the first time in this illness, which kills 50 percent of patients in less than a year. Furthermore, about a fifth of individuals who received the therapy were alive four years after treatment, at which point everyone on the control regimen had died. Although side effects were significant, most could be managed.
The US Food and Drug Administration (FDA) approved ipilimumab for late-stage melanoma in 2011. According to data published earlier this year, more than 20 percent of 5000 individuals treated with the antibody were still alive ten years after treatment.
Releasing additional brakes
In the meantime, a related story was unfolding. Tasuku Honjo (Kyoto University) discovered a second protein, PD-1, that also obstructs T-cell activation, but through a different pathway, according to subsequent work by him in collaboration with Gordon Freeman (Dana-Farber Cancer Institute). Mice that lack PD-1 remain healthy until adulthood, and only then develop autoimmune symptoms. These observations and others suggested that anti-PD-1 treatment would melt tumors while eliciting fewer side effects than anti-CTLA-4.
Medarex developed therapeutic antibodies that target either PD-1 or the protein partner that stimulates it, PD-L1. In 2012, a group led by Suzanne Topalian (Johns Hopkins University School of Medicine) reported that anti-PD-1 benefits individuals with non-small-cell lung cancer, melanoma, and renal-cell cancer, thus confirming the notion that a single drug that boosts the immune system can combat different tumors. The FDA approved two anti-PD-1 antibodies (nivolumab/Bristol-Myers Squibb and pembrolizumab/Merck & Co.) for metastatic melanoma in 2014, and it extended approval to metastatic squamous non-small cell lung cancer earlier this year.
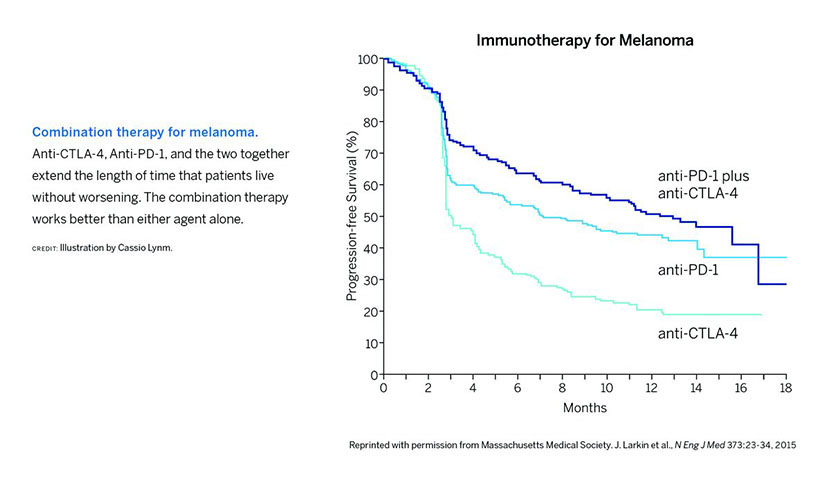
Scientists are developing additional therapies to impede more than a dozen other proteins that normally curb immune responses. Greater than 500 clinical trials are under way to explore whether these agents can defeat many types of malignancies, including ovarian cancer, bladder cancer, head and neck cancer, and gastric cancer. Combination regimens are particularly impressive. For example, anti-CTLA-4 and anti-PD-1 together perform better than either antibody alone (see Figure). Preliminary results suggest that about 80 percent of people with advanced melanoma remain alive two years after treatment — even though the disease in many of these patients initially progressed or seemed to progress.
Allison’s work has cracked open a brand new therapeutic world that capitalizes on the body’s inherent capacity to quash malignant cells. His discoveries are transforming cancer treatment and the prognosis for individuals with these life-threatening diseases.
by Evelyn Strauss
Key publications of James Allison
Leach, D.R., Krummel, M.F., and Allison, J.P. (1996). Enhancement of antitumor immunity by CTLA-4 blockade. Science. 271, 1734-1736.
Van Elsas, A., Hurwitz, A.A., and Allison, J.P. (1999). Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 190, 355-366.
Quezada, S.A., Peggs, K.S., Curran, M.A., and Allison, J.P. (2006). CTLA-4-blockade and GMCSF combination immunotherapy increases effector frequencies altering the intra-tumor balance of effector and regulatory T cells. J. Clin. Invest. 7, 1935-1945.
Curran, M., Montalvo, W., Yagita, H., and Allison, J.P. (2010). PD-1 and CTLA-4 combination blockade expands infiltrating T-cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA. 107, 4275-4280.
Wolchok, J.D., Hodi, F.S., Weber, J.S., Allison, J.P., Urba, W.J., Robert, C., O’Day, S.J., Hoos, A., Humphrey, R., Berman, D.M., Lonberg, N., and Korman, A.J. (2013). Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. N.Y. Acad. Sci. 1219, 1-13.
Sharma, P. and Allison, J.P. (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 161, 205-214.
Sharma, P. and Allison, J.P. (2015). The future of immune checkpoint therapy. Science.348, 56-61.
Award presentation by Michael Bishop
In 1890, the Russian playwright and physician Anton Chekov wrote a letter in which he alluded to a centuries-old observation that infections seemed to slow the growth of malignant tumors. For Chekov and most others of his time, the observation held little meaning. For William Coley, however, it fueled a medical crusade.
Coley was a contemporary of Chekhov’s and a bone surgeon at what is now the Memorial Sloan Kettering Cancer Center. Inspired by the death of one of his earliest patients, he reviewed the case histories of individuals with bone cancer and noticed that bacterial infections following surgery seemed to improve the outcome. He reasoned that infection stimulated the immune system, which in turn destroyed cancer cells that had escaped the surgeon’s knife.
Acceptance remarks by James P. Allison
Interview with James Allison
Video Credit: Flora Lichtman
