
Kazutoshi Mori
Kyoto University

Peter Walter
University of California, San Francisco
The 2014 Albert Lasker Basic Medical Research Award honors two scientists for their discoveries concerning the unfolded protein response, an intracellular quality-control system that detects harmful misfolded proteins in the endoplasmic reticulum and signals the nucleus to carry out corrective measures. Kazutoshi Mori (Kyoto University) and Peter Walter (University of California, San Francisco) identified core components of this process and unveiled unexpected aspects of its mechanism.
Approximately one-third of cellular proteins pass through the endoplasmic reticulum (ER), a netlike labyrinth of membrane-bound tubes and flattened sacs inside the cell. Work in the 1960s revealed that the ER sorts and transports proteins that are destined for export or the cell’s surface, and we now know that the ER allows cargo to pass only after applying stringent standards. In particular, proteins must assume correct three-dimensional shapes to perform their jobs, and the ER fosters this outcome. Furthermore, when unfolded proteins accumulate in this compartment, the cell bolsters the ER’s folding capacity. This phenomenon forms the linchpin of the unfolded protein response (UPR).
Three years later, Hugh Pelham (Medical Research Council, Cambridge) established that one of the GRPs, GRP78, resides in the ER and resembles a protein that prevents heat-damaged proteins from clumping. When glucose supplies drop, sugars that normally decorate some proteins are no longer available. Pelham proposed that the resulting sugar-deficient proteins stick together, perhaps because they misfold, and that GRP78, like its molecular relative, thwarts protein aggregation. Pelham also found that GRP78 was identical to another protein, BiP, that associates with partially assembled antibody molecules in the ER. In parallel, Mary-Jane Gething and Joseph Sambrook (University of Texas Southwestern Medical Center) showed that BiP attaches to misfolded forms of a different protein in the ER.
These findings hinted that BiP helps proteins fold; if true, manufacture of BiP in response to unfolded proteins would serve a clear benefit. The connection between glucose starvation and folding remained murky, however, and the model relied on that link. In 1988, Gething and Sambrook established that misfolded protein rather than sugar-adornment defects sends the alert to ramp up BiP output.
In 1989, yeast BiP surfaced. Its quantities also climb in response to unfolded ER proteins. Mori joined Gething and Sambrook’s lab as a postdoctoral fellow, and the group identified a short series of DNA letters that abuts the BiP gene. This sequence spurs molecular machinery to copy, or transcribe, the BiP DNA into mRNA when unfolded proteins accumulate in the ER; the sequence, when placed next to a different gene, similarly turns on its transcription.
Together, these observations suggested that cells must somehow monitor the abundance of unfolded proteins in the ER and transmit that information to the nucleus, which houses the genes. These events spark production of BiP and other proteins that promote folding, which reverse the problem. But no one knew how the nuclear equipment senses the ER environment.
The complexity of Ire1
Independently, Mori, in Texas, and Walter, in San Francisco, placed the DNA stretch that Mori had uncovered next to a gene whose product makes a blue substance. When unfolded proteins accumulate in the ER and the engineered yeast cells send the usual signal to the nucleus, it stimulates not only typical UPR targets, but also the gene that turns the yeast blue. Yeast with defects in the UPR system would not change color, the researchers reasoned.
In 1993, the investigators used this scheme to isolate white yeast strains and thus tracked down the faulty genes whose normal versions presumably contribute to the UPR. One encodes a protein called Ire1.
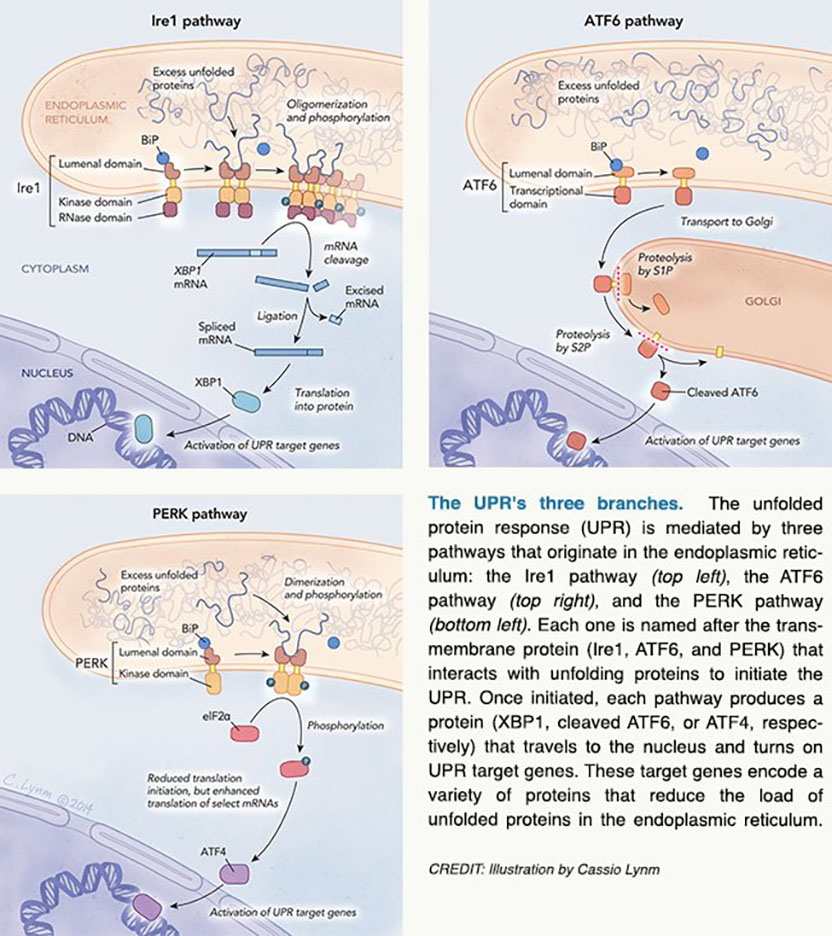
Sequence analysis of Ire1 suggested that it is a kinase — an enzyme that adds phosphates to itself and/or other proteins. Additional work by Walter and Mori confirmed and extended this prediction. They found that Ire1 lies in the ER membrane with its kinase portion in the cytoplasm. In this orientation, the ER region could detect an unfolded protein signal and the other end could convey the message to cytoplasmic partners.
Mammalian kinases were well known to monitor environmental cues and, by adding phosphates to themselves or other molecules, trigger adaptive physiological changes. Perhaps, Mori and Walter reasoned, Ire1 would behave similarly.
To figure out how Ire1 delivers the unfolded-protein message, Walter and Mori (by then an independent investigator in Japan) set out to identify the presumptive courier that picks up the signal and carries it to the nucleus. They sought a protein that binds to the DNA sequences adjacent to UPR target genes and provokes transcription. The investigators captured the component they sought, a protein that previously had been named Hac1.
Their results, reported in 1996, contradicted expectation. In the simplest scenario, the theoretical protein to which Ire1 affixes a phosphate would be ready for action upon stimulation. Hac1, however, is not ready for anything; rather, it is manufactured only after the UPR alarm rings.
A crucial clue to explain this result came from the observation that the Hac1-encoding mRNA shrinks when unfolded proteins accumulate. Instead of adding a phosphate to another protein, Ire1 prompts removal of a chunk of Hac1’s mRNA. Additional work by Walter, which was confirmed and extended by Mori, established that HAC1 precursor mRNA contains an internal stretch of 252 genetic letters that is eliminated to supply the blueprint for active Hac1.
A canonical molecular machine splices sequences from precursor mRNAs and operates in the nucleus. The plot thickened when Walter showed that this apparatus does not act on HAC1 mRNA. Instead, he found, the severed HAC1 mRNA is stitched together by a cytoplasmic enzyme — tRNA ligase — that normally joins the two components of a different type of RNA, transfer RNA.
The search was now on for an enzyme that excises the middle piece of the HAC1 precursor mRNA. Inspired by a related protein’s behavior, Walter showed that the cytoplasmic segment of Ire1, which contains the kinase and an additional stretch of protein, could cut HAC1 precursor mRNA at the expected sites. Then he demonstrated that the splicing reaction could occur in the test tube with only two enzymes: Ire1 cleaves the HAC1 precursor mRNA at both splice junctions, and the transfer RNA ligase sews them together.
Mammalian systems unfold
As these details of the yeast UPR were materializing, researchers were struggling to gain traction in the mammalian system. In 1998, Mori unearthed a sequence that was common only to genes that fire up in response to unfolded ER proteins. This element rouses several UPR target genes, he found. Furthermore, a human protein called ATF6 binds to this DNA motif and activates adjacent genes.
Mori noticed that an overabundance of unfolded proteins incites conversion of full-length ATF6 to a smaller version; the large form dwells in the ER, whereas the trimmed one resides in the nucleus. This and other work suggested that excess unfolded proteins trigger release of a portion of ER membrane-bound ATF6. The liberated fragment travels to the nucleus and activates transcription of UPR target genes.
While Mori was discovering and elucidating ATF6’s role in the UPR, David Ron (New York University School of Medicine) and Randal Kaufman (University of Michigan Medical Center) found mammalian versions of Ire1, which share fundamental functional features with their yeast cousin. Three years later, Mori and Ron identified the human and worm versions of yeast Hac1, a protein known as XBP1.
In the meantime, near the beginning of 1999, David Ron and Ron Wek (Indiana University School of Medicine) had independently uncovered a third arm of the UPR, which depends on a protein called PERK. Like Ire1 and ATF6, PERK also lies across the ER membrane. Furthermore, its ER domain resembles that of Ire1. On the cytoplasmic side, a protein kinase segment of PERK adds phosphates to a particular protein, which then impedes translation of mRNAs. As a result, fewer proteins enter the ER, thus lightening the folding load.
Strength in numbers
In the last ten years, Walter, with UCSF colleague Robert Stroud, has peered more closely at Ire1 activation with X-ray crystallography. Previous work by Mori, Walter, and others had suggested that UPR induction causes Ire1 molecules to snuggle up in the membrane. By studying yeast Ire1, Walter and Stroud provided an atomic-level rationale for those results and illuminated details of the reaction.
In addition to providing assistance during protein folding, BiP attaches to Ire1 on the side that lies within the ER; when BiP falls off, naked Ire1 molecules pair up and create grooves that bind the unfolded proteins, Walter and Stroud suggest. Multiple Ire1 duos then congregate to form higher order structures; such association rearranges their cytoplasmic segments, positioning them so they can grab and then snip the HAC1/XBP1 mRNA, according to the model.
Researchers are still uncovering layers in the UPR. For example, Ire1 governs ER membrane synthesis and a system that shuttles recalcitrant unfolded proteins from the ER to a cellular incinerator. Even with these additional components, the unfolded protein burden sometimes surpasses the cell’s management capacity. That situation can trigger cell suicide, which obliterates unhealthy cells that might otherwise wreak havoc. Investigators are deciphering how the Ire1, ATF6, and PERK branches of the pathway help cells make life-and-death decisions.
Many scientists are now pursuing ways to harness the UPR for medical advantage. Certain forms of some inherited diseases that cause elevated cholesterol levels, cystic fibrosis, and retinitis pigmentosa produce abnormal proteins that do not fold properly and overwhelm the UPR.
Walter and Mori have unraveled a process with numerous unusual features. Their work has unlocked a multi-layered, highly choreographed system that lies at the heart of normal cellular function.
by Evelyn Strauss
Key publications of Kazutoshi Mori
Mori, K., Ma, W., Gething, M.J., and Sambrook, J.F. (1993). A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 74, 743-756.
Mori, K., Kawahara, T., Yoshida, H., Yanagi, H., and Yura, T. (1996). Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells. 1, 803-817.
Kawahara, T., Yanagi, H., Yura, T., and Mori, K. (1997). Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell. 8,1845-1862.
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 10, 3787-3799.
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 107, 881-891.
Sato, Y., Nadanaka, S., Okada, T., Okawa, K., and Mori, K. (2011). Luminal domain of ATF6 alone is sufficient for sensing endoplasmic reticulum stress and subsequent transport to the Golgi apparatus. Cell Struct. Funct. 36, 35-47.
Key publications of Peter Walter
Cox, J.S., Shamu, C.E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 73,1197-1206.
Cox, J.S. and Walter, P. (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 87, 391-404.
Sidrauski, C. and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 90, 1031-1039.
Gonzalez, T.N., Sidrauski, C., Dörfler, S., and Walter, P. (1999). Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J. 18, 3119-3132.
Korennykh, A.V., Egea, P.F., Korostelev, A.A., Finer-Moore, J., Zhang, C., Shokat, K.M., Stroud, R.M., and Walter, P. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature. 457, 687-693.
Gardner, B.M., Pincus, D., Gotthardt, K., Gallagher, C.M., and Walter, P. (2013). Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5, a013169.
Award presentation by Michael Brown
 Henry Ford is credited with inventing the assembly line, but Mother Nature beat him by a billion years when she invented the endoplasmic reticulum, Mother Nature’s assembly line. In Ford’s assembly line parts are passed from one worker to another. At the end of the line is the inspector, who tests the product. Correctly assembled products are exported. Defective products are destroyed. If too many products are defective, the inspector sends a signal to the control room, where supervisors dispatch troubleshooters to correct the problem.
Henry Ford is credited with inventing the assembly line, but Mother Nature beat him by a billion years when she invented the endoplasmic reticulum, Mother Nature’s assembly line. In Ford’s assembly line parts are passed from one worker to another. At the end of the line is the inspector, who tests the product. Correctly assembled products are exported. Defective products are destroyed. If too many products are defective, the inspector sends a signal to the control room, where supervisors dispatch troubleshooters to correct the problem.
Ford never knew he was copying the endoplasmic reticulum, which I will abbreviate as ER. The ER is a network of membrane-bound tunnels within every nucleated cell from yeast to human. Like Ford’s assembly line, the ER makes products for export. Its products are proteins designed to function not in the controlled environment of the cell, but in the harsh environment outside. Some of the proteins are destined to reside on the cell surface, where they serve as receptors that respond to external signals. Other proteins are secreted to influence the behavior of other cells. Lasker laureate Günter Blobel showed that exported proteins are inserted into the ER during their translation. Once inside, they are bound by the factory workers of the ER, which are proteins called chaperones. Like Ford’s welders, the chaperones help the exported proteins to fold into their final three-dimensional structures. The ER assembly line must perform perfectly. Mistakes are not tolerated. Unlike in the auto industry, in biology there are no recalls.
Acceptance remarks

Acceptance remarks, 2014 Lasker Awards Ceremony
It is my very great pleasure and honor to receive the Lasker Award. Looking back, I can point to many happy meetings that changed the directions of my career. In particular, three meetings come to mind. The first happy meeting took me to the United States. I did not like biology in high school. Textbooks seemed descriptive. I felt I had to learn everything by heart. As a freshman of Kyoto University, most of my learning came from newspapers. I was amazed by the fact that the genetic code is conserved from Escherichia coli to humans, which allowed scientists to produce human proteins in E. coli. I hoped for a bright future in biology. So, I switched my major from chemistry to biology.
Since there was no molecular biology program at my school, I studied biochemistry. After graduating, I obtained a permanent position at a local university. This was very fortunate because Japan had no post-doc system at that time. I worked hard but did not enjoy the biochemical project I pursued — I wanted to do something more interesting and important. I decided to quit the position and go to United States, where I had my first happy meeting: in 1989, at the University of Texas Southwestern Medical Center, I met the discipline of molecular biology under the mentorship of Mary-Jane Gething and Joe Sambrook.
There, in Dallas,Texas, I was introduced to the unfolded protein response (UPR) — my second happy meeting. I succeeded in cloning IRE1, which turned out to be a key molecule to unraveling the UPR in yeast. To my surprise, Peter Walter came up with the same molecule at the same time. In our first papers, published back to back in Cell, Peter and I discovered that IRE1 had a strong kinase activity.
After four-and-a-half years in Texas, I came back to Japan and found a position at the HSP Research Institute in Kyoto. My next targets were transcription factors specific to yeast UPR. My new director advised that I be careful: IRE1 is a kinase, he said, and there may be a downstream kinase cascade. So, you might end up obtaining many kinases but no transcription factor. And because the Institute focuses on transcription, we do not want kinases. He told me to think of a method of obtaining a transcription factor directly. But this was a difficult task. After a 14-month struggle, I finally came up with a simple method called one-hybrid screening, which turned out to be the right approach. It worked magnificently, and in 1996, I identified HAC1, the key transcription factor for the UPR In yeast. And with my one-hybrid screening approach, several years later my colleagues and I obtained two other crucial transcription factors for the UPR in human and animal cells.
Had I stayed in the United States after my Texas days, I would have probably employed a complicated American-style screening approach that would have never allowed me to keep up with a major player like Peter Walter. But the development of my simple one-hybrid screening was my third happy meeting — to me, it was the Japanese version of the American dream. Thank you again for honoring me with the Lasker Award.

Acceptance remarks, 2014 Lasker Awards Ceremony
Four years ago, Joe Goldstein issued the ultimate instructions on “How to Win a Lasker.” In his short piece in Nature Medicine, he presents a list of advice, “culled from the experts,” as he puts it, and then promptly dismisses it as a “not foolproof formula.” He continues to quote as better advice the British mathematician Godfrey Hardy, who defined in the early 1900s scientific beauty as an art form in which outstanding science gives you “cerebral chills and intellectual kicks” — that combine the qualities of significance, generality, and unexpectedness.
Biology, however, is not mathematics, nor is it physics, nor chemistry. As biologists, we are not free to impose our own axioms to inject beauty into our work or even to assume elegant adherence to deterministic logic. In biology, Mother Nature presents the playing field, and it is our task to decipher how it works. Disconcertingly, Nature deploys the strategy of random walk, of mutation and selection, leading to the evolution of the world that surrounds us. She then presents us with the most fascinating puzzles to decipher: the inherently unpredictable Rube Goldberg machines that make up a living cell.
When we began working on the unfolded protein response that we celebrate here today, we found ourselves traveling along a road that seemed comfortable and predictable, perhaps too narrow and straight. We asked but simple questions: “how does one part of the cell know what is happening in another?” And, we got a simple answer: a kinase receptor that, like many others, controls the cell’s gene expression system. But suddenly, the ground shifted, and we stepped into a morass, in which the seemingly familiar dots that before gave us a sense of security no longer connected. Walking on, undeterred, we deciphered one of most unusual cell-internal communication pathways, made up almost entirely from an unprecedented potpourri of repurposed components. To top it off, the salient features of what we learned from simple, single-celled brewer’s yeast holds true for our own cells, and these features now emerge as impacting players in a plethora of human diseases — giving us hope that our esoteric findings will be translated one day into tangible benefits for mankind.
My main point here is that none of this was predictable. Neither I nor Kazu stand here today as genius mathematicians akin to Hardy or artists, who begin with an empty canvas and control over the elegance of their work. We are explorers — not designers — facing the chaotic randomness of evolution. We diligently deciphered one of Nature’s guarded mysteries, and only then could we condense our findings to their most elementary beauty. To us, it has been a fantastic journey filled with adventure, cerebral chills, and intellectual kicks. Yes, this journey required radical thinking and fearless experimentation. But only in the end, did it all combine — serendipitously — into a story of unexpectedness and, we hope, lasting significance and generality.
We are thrilled to be here today, and we deeply appreciate that our work has been so well received.
Thank you for such a nice celebration.
A toast to discovery!
Interview with Kazutoshi Mori and Peter Walter
Video Credit: Susan Hadary
